2 Factors Affecting Drug Disposition
Ideally, fixed dosing regimens are based on scientific studies performed with the drug of interest in the target species. Often, however, the dose for a drug used in dogs or cats is extrapolated from other species, especially humans to dogs and humans or dogs to cats, rather than based on scientific studies. For those drugs backed by scientific evidence, sample numbers are often too small, resulting in marked variability in the pharmacokinetic parameters. Further, as in human medicine, animals in which pharmacokinetic studies are performed are generally healthy and may not represent the state of disease in the animals treated with the drug. A number of factors can alter plasma drug concentrations (PDCs) in the patient due to changes in drug disposition, thereby increasing the risk of therapeutic failure. Many of these can be anticipated, allowing for adjustments in the dosing regimen. In addition to the disposition of a drug, these factors might also alter patient response to the drug. These factors might be categorized as physiologic factors such as species and age; pathologic factors, particularly cardiac, renal, or hepatic disease; and pharmacologic factors that occur when one drug alters the kinetics or response to another drug (Box 2-1).
Box 2-1 Examples of Factors that Affect Drug Disposition
Physiologic Factors
Age-Induced Differences
Drug Disposition in the Geriatric Animal
This discussion focuses on some of the clinically important changes that are likely to alter response to drug therapy in geriatric patients (Table 2-1) and on actions that can be implemented to compensate or reduce the sequelae of these changes. Age is associated with changes both in pharmacokinetics (gerontokinetics) and pharmacodynamics, with changes generally compared with those of the young adult.1–3 Data in animals are limited, but in general, disposition pattern changes described for humans appear to extrapolate adequately to dogs and cats.4 Geriatric animals are at a greater risk for therapeutic failure because of normal aging changes in physiology, an increased incidence of disease, and the likelihood of polypharmacy in response to disease; moreover, a decrease in normal organ protective mechanisms may increase the risk of adverse events. The age at which body functions shift from a period of growth to a period of decay (16 to 18 years in humans) has not been established in dogs and cats. The age probably differs among canine breeds. Aging is accompanied by permanent loss of up to 30% of body cells, with a parallel loss in oxygen consumption. Body composition changes, as do regional blood flow rates. Physiologic functions generally decline steadily with increasing age. In humans basal metabolic rate decreases by 0.4% per year2; according to the National Research Council,5 energy requirements of dogs decrease as they age. Changes among the body systems can influence all four drug movements.
Cardiovascular
As animals age, cardiac output decreases and circulation transit time increases. In humans cardiac output decreases by about 1% per year for a total decline of 30% to 40% in the aged. Regional and organ blood flow rates similarly decrease.2 The net effect of these changes depends on the state of disease but can influence each drug movement. Absorption, metabolism, and excretion are likely to decrease, whereas distribution may increase or decrease depending on the state of vascular responses or fluid retention.2,6,7 As cardiac function decreases, secondary compensatory responses can lead to further risks of adverse reaction.7 Blood flow is preferably redistributed to the brain and heart, increasing the risk of toxicity of drugs toxic to these tissues.
Central and peripheral nervous systems
As the geriatric patient ages, brain weight and peripheral fiber numbers decrease. Connective tissue infiltrates peripherally.2 Oxygen consumption and cerebral blood flow decrease. In addition, decreased amounts of selected neurotransmitters have been documented. Morita and coworkers8 reported alterations in the blood–brain barrier in elderly dogs.
Respiratory
In human geriatric patients, residual lung volume decreases by 50% with accompanying decreases in vital capacity, arterial oxygen pressure (PO2), and maximum oxygen uptake. In addition, the central response to hypoxia and hypercapnia, such as that induced by opioid analgesics, decreases.2 Anesthetic or other sedating agents must be used more cautiously.
Gastrointestinal
As animals age, deglutition decreases as a result of decreased salivation and pharyngeal and esophageal motility. Gastric function is characterized by atrophy of the mucosa with a reduction of hydrochloric acid secretion and a subsequent increase in gastric pH. Gastrointestinal motility is generally reduced. The intestinal macrovilli and microvilli also atrophy, increasing the risk of bacterial overgrowth. These sequelae tend to reduce the absorption and thus the PDC of orally administered drugs. Changes in gastrointestinal function (including reduction of gastroprotective effects) may also predispose the geriatric patient to adverse effects induced by toxic drugs such as chemotherapeutic agents and nonsteroidal antiinflammatory drug (NSAID) analgesics. The latter should be used with caution; the clinician should anticipate and be prepared to treat toxicities. Digestibility of key nutrients, such as fat, tends to decrease in elderly cats.9 As with humans, the intestinal microbiota population shifts in elderly dogs and cats, with lactobacilli decreasing and clostridia increasing in dogs10 and bifidobacteria decreasing in cats.11 In addition to response to antimicrobials, these changes might influence enterohepatic circulation of bile acids and thus, potentially, some drugs.12
Hepatic
Changes in hepatic function are important to the geriatric animal because of the liver’s role in the metabolism of drugs.13 Hepatocyte number and function decrease, as do hepatic and splanchnic blood flow, hepatic oxidation, and cytochrome P450 content (the primary drug-metabolizing enzyme). Both flow-limited and capacity-limited drugs are affected. For example, hepatic clearance of both opioid analgesics (which are characterized by first-pass metabolism; i.e., flow limited) and nonsteroidal analgesics (eliminated principally by hepatic metabolism; i.e., capacity limited) is decreased in geriatric patients. Increased response of human geriatric patients to opioid analgesics—they require 60% to 75% less drug than younger patients do—has been attributed to changes in drug elimination.14,15 Changes in hepatic function, oxygenation, and nutrition may also predispose the liver to drug-induced hepatotoxicity. Because of reduced hepatic function, the geriatric patient may be less able to generate endogenous hepatoprotectant agents, increasing the risk of drug-induced hepatotoxicity (Chapter 4).
Body weight and composition
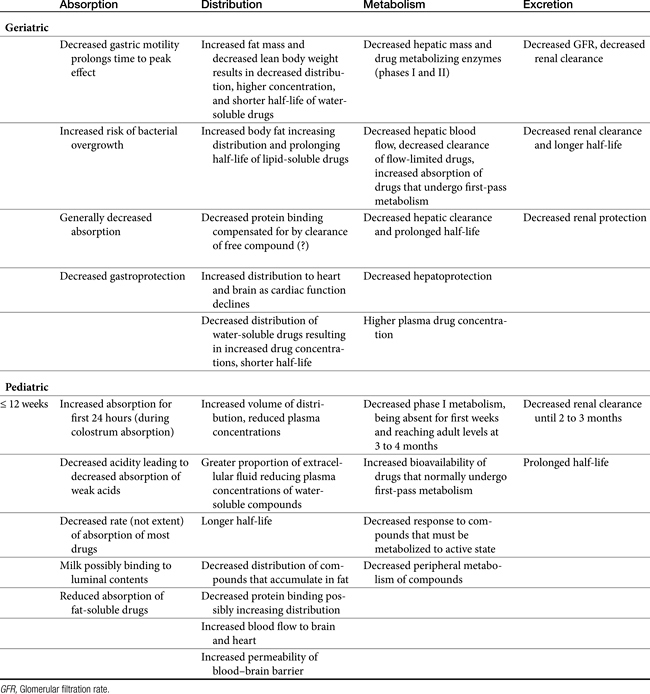
Changes in body composition may be among the most complex in the geriatric animal. Like humans, dogs tend to lose lean body mass and accumulate body fat as they age (Figure 2-1).16 In male humans, fat increases from approximately 18% in young adults up to 50% in the aged.2 Increased proportion of body fat is accompanied by a decrease in total body water and cell mass. Although extracellular fluid does not change in total amount, the relative proportion of total body water that it makes up increases. Thus the proportion of intracellular to extracellular fluid decreases. The sequelae of these changes depend on the drug. As distribution of water-soluble drugs decreases with total body water, PDCs tend to increase. The distribution of lipid-soluble drugs increases as the proportion of body fat increases, however, which tends to decrease PDCs unless the patient is dosed on a mg/kg basis. The impact of aging on body composition in dogs varies with breeds. For example, in one study body fat increased with age in Great Danes but not Labrador Retrievers or Papillons,17 although this did occur within 1 year of death in elderly Labrador Retrievers in another study.16 Although middle-aged cats tend to be overweight, geriatric cats often become thin.9 Thus PDCs of water-soluble compounds might be expected to be higher in older animals compared with those of young adults, even if dosing on an mg/kg body weight basis, whereas for lipid soluble drugs, dosing on an mg/kg body weight basis might compensate for potential changes in plasma concentrations.
Receptor sensitivity and pharmacodynamics
Geriatric patients respond differently to some drugs, which suggests that tissue receptor sensitivity to the drugs is altered. Changes in receptor number or responsiveness have been implicated but not documented.2,3 Physiologic changes such as altered neurotransmission or intracellular constituents have also been suggested. For example, geriatric patients are less likely to perceive, appreciate, or express pain. Thus the need for analgesic therapy is often not detected. In addition, geriatric patients are less able to respond to many analgesic drugs.
Disease
Aged animals are more likely to be suffering from diseases that affect not only drug disposition but also tissue receptivity to drugs and organ protection.15 The immune system of the geriatric patient is not as effective as that of the adult,18 leading to the use of bactericidal antimicrobials and minimizing the use of immunosuppressive drugs. In addition, the geriatric patient is more likely to be receiving multiple drugs, which increases the likelihood of drug interactions. Finally, diseases of selected organs may predispose these organs to drug-induced toxicity.
Drug Disposition in the Pediatric Animal
With regard to dogs and cats, pediatric generally refers to the first 12 weeks of life.19 Important developmental changes occurring within this time spectrum, however, justify further staging into neonatal (0 to 2 weeks), infant (>2 to 6 weeks), and pediatric (>6 to 12 weeks) periods of growth. Changes associated with each of these periods cause accompanying changes in drug disposition, thus rendering the pediatric patient more susceptible to drug-induced adverse reactions. All four determinants of drug disposition (i.e., absorption, distribution, metabolism, and excretion) undergo dramatic changes as the neonate matures (see Table 2-1).20,21 However, the clinical significance of these sequelae varies.
KEY POINT 2-2
Drug clearance generally does not reach adult capacity until approximately 3 months of age.
Absorption
Young kittens have decreased energy, carbohydrate, and organic matter digestibility compared with kittens older than 19 weeks of age.22 However, because the surface area of the small intestine is large, even in neonates, the extent of drug absorption probably does not clinically differ between normal pediatric and adult animals. The rate of absorption tends to be slower in pediatric animals, however, probably because of decreased gastric emptying and irregular intestinal peristalsis. As a result, peak PDCs may be lower in pediatric patients. The decreased rate of absorption might actually protect against toxic drug concentrations.23,24 However, protection may not be present before absorption of colostrum. During this period the permeability of the intestinal mucosa is increased, leading to increased rate and extent of drug absorption. Occasionally, drugs that normally are not absorbed from the gastrointestinal tract (e.g., aminoglycosides, carbenicillin, and other acid-sensitive beta-lactams and enteric sulfonamides) can reach systemic circulation. Intestinal permeability decreases rapidly after the ingestion of colostrum,24,25 possibly because of the endogenous release of hydrocortisone or adrenocorticotropic hormone. Exogenous supplementation of either of these hormones by the 24-hour prepartum mother prevents increased permeability and colostrum absorption in the neonate.
A number of other factors may alter small intestinal drug absorption in pediatric patients. Gastric pH is neutral in the newborn; adult levels are not reached until sometime after birth, depending on the species.23,25 Increased gastric pH (achlorhydria) may decrease the absorption of many drugs that require disintegration and dissolution or are ionized in a less acidic environment (e.g., weak acids such as penicillins). Milk diets can reduce drug absorption by either decreasing gastric emptying or directly interacting with drugs (e.g., milk can impair absorption of tetracyclines). The “unstirred water layer” adjacent to the surface area of the mucosal cells is thicker in the neonate than in the older pediatric patient and may limit the rate of absorption of some drugs. As biliary function matures, the absorption of fat-soluble drugs (e.g., griseofulvin and fat-soluble vitamins) increases. Microbial colonization of the gastrointestinal tract may alter response to antimicrobial drugs, extrahepatic metabolism, or enterohepatic circulation.26,27
Absorption from the rectal mucosa is rapid. Rectal administration of drugs or fluids can be used for pediatric patients when venous catheterization is difficult, to reduce complications associated with intravenous administration (e.g., sedation, anesthesia), or when oral administration is undesirable (e.g., antiemetics). Several pediatric drugs intended for systemic effects are available as rectal suppositories. Limited data from studies of human infants indicate that peak plasma concentrations after rectal administration may be higher than those obtained by other routes.26
Absorption of drugs administered parenterally to pediatric animals also varies from that of adults. The rate of absorption after intramuscular administration changes with age as muscle mass and its accompanying blood flow increase and as vasomotor responses mature.26 Because muscle mass is small, subcutaneous administration is frequently preferred for pediatric patients. Again, variability in subcutaneous absorption rates can be anticipated with age. Less fat but greater water may result in faster absorption compared with that in adults.28 Environmental temperature probably influences subcutaneous absorption, particularly in newborns whose thermoregulatory mechanism functions poorly. Cold environments are likely to reduce subcutaneous drug absorption if the neonate is not kept warm. The same is true for patients in a state of hypothermia. Intraperitoneal administration can be a lifesaving route of blood and fluid administration, particularly for the newborn with inaccessible central veins. Isotonic fluids are rapidly absorbed, and up to 70% of red blood cells are absorbed in 48 to 72 hours.29 Blood and fluids can also be administered into the medullary cavity of large bones.30,31
Absorption of volatile anesthetics from the pediatric respiratory tract is rapid because minute ventilation is greater.19 Thus young animals are more sensitive to the effects of gas anesthetics. Although not a common route of drug administration, percutaneous absorption of drugs is likely to be greater in pediatric patients. Percutaneous absorption is directly related to skin hydration, which is greatest in neonates. Topical administration of potentially toxic lipid-soluble drugs (e.g., hexachlorophene and organophosphates) is not recommended.
Distribution
The most important factors contributing to differences in drug distribution in pediatric patients are differences in body fluid compartments and drug binding to serum proteins. Body fluid compartments undergo profound changes with the growth of the neonate. Both the percentage of total body water and the ratio of compartmental volumes change with maturation. The percentage of total body water decreases with age, but the decrease is more substantial in the extracellular versus the intracellular compartment (see Table 2-1; Figure 2-2).32 Daily fluid requirements are greater in neonatal and pediatric patients, in part because a larger proportion of their body weight is represented by body water (see Figure 2-2). The sequelae of these body compartment differences depend on the normal distribution of the drug. Most water-soluble drugs are distributed to extracellular fluids. In pediatric patients the volume to which these drugs is distributed is therefore higher than in adults; PDCs correspondingly decrease. Thus it may be necessary to increase doses to prevent therapeutic failure. A different pattern might be expected for lipid-soluble drugs because they tend to be distributed to total body water. Such drugs should be dosed according to body weight (e.g., mg/kg). Although decreased PDCs resulting from increased distribution may protect the pediatric patient from potentially toxic drug concentrations,33 a poor therapeutic response may result from failure to generate therapeutic drug concentrations of water-soluble drugs. The disposition of several antimicrobials in neonatal animals has been reviewed by Baggott,34 with differences in disposition generally found to be similar to those described for humans. Ampicillin is characterized by volumes that are larger in puppies and kittens (threefold to fourfold higher) compared with adults, resulting in clinically significant lower drug concentrations.35,36 Distribution of enrofloxacin, a lipid-soluble drug, is somewhat unpredictable and age dependent in kittens, being smaller at 2 to 4 weeks but greater at 4 to 6 weeks.37 Changes in the half-life of each drug parallel changes in distribution. Because many drugs are distributed to a larger volume in pediatric patients, a longer half-life should be anticipated, and it may be necessary to prolong the dosing interval. However, for enrofloxacin in kittens this effect was balanced by increased clearance at 6 weeks.37

Because the proportion of body fat is smaller in pediatric patients, the distribution of lipid-soluble drugs that accumulate in fat (e.g., organophosphates, chlorinated hydrocarbons, ultrashort thiobarbiturates) may be proportionately decreased. Although drug half-life would decrease, PDCs may become toxic. Many lipid-soluble drugs have a high affinity for and are bound by plasma proteins, thus facilitating their movement through the body. Binding, however, limits their distribution to tissues. Predicting the distribution of highly protein-bound drugs is complicated in the pediatric patient. Serum concentrations of both serum albumin, the protein to which most drugs are bound, and α1-glycoproteins (to which basic drugs preferentially bind) are decreased in pediatric patients.38 Protein binding of drugs may also be reduced because of differences in albumin structure or because drugs compete with endogenous substrates (e.g., bilirubin) for binding sites.24,39 As drugs are displaced, the concentration of free, pharmacologically active drugs and the risk of adverse reactions increases. These changes are significant, however, only if the drug is highly (i.e., >80%) protein bound and characterized by a small therapeutic index. Although the concentration of free drug increases, that of total drug in the plasma tends to decrease because unbound drug is free to distribute into tissue.39 Consequently, drug half-life may increase, and longer dosing intervals may be indicated for potentially toxic drugs. Increased clearance of unbound drug may ultimately normalize a half-life that has been lengthened by an increased volume of distribution.
Differences in regional organ blood flow might cause clinically important changes in drug disposition in pediatric animals. Differences in renal blood flow have been documented40,41 and result in clinically important differences in drug excretion. Blood flow to vessel-rich tissues of the body (i.e., heart and brain) is greater and faster;19 the pediatric patient is thus more susceptible to drug-induced cardiac and central nervous system (CNS) toxicity. The potential for CNS toxicity is further increased because the blood–brain barrier is poorly developed immediately after birth. Increased permeability protects the neonatal brain from a deficiency of nutritional fuels in stressful states (e.g., hypoglycemia, hypoxia, acidosis) by allowing the movement of oxidizable substrates such as lactate into brain cells.42 The status of efflux proteins is not yet established. Drugs normally incapable of reaching the adult brain are, however, also able to reach brain cells, which are very susceptible to their effects, thus increasing the risk of CNS toxicity.43,44
Metabolism
Drug elimination, including both hepatic metabolism and renal excretion, is limited in neonatal and pediatric patients. Thus many drugs administered to the young animal are characterized by decreased clearance.20,24 In contrast to human infants, hepatic metabolism of drugs is incompetent in the near-term and neonatal puppy.45–47 Both phase I (e.g., oxidative) and phase II (e.g., glucuronidation) reactions are reduced. The various pathways of metabolism mature at different rates. Phase I activity may not occur in the neonatal puppy and may not be evident until day 9 if it does. Activity appears to progressively increase after day 25, not reaching adult levels until 135 days postpartum.46 Drug metabolism in pediatric patients, however, is quite complex. For example, Ecobichon and coworkers48 found that in Beagle puppies, phase I metabolites decreased with age, and for phase II metabolism, glucuronidation was the most predominant, with sulfonation decreasing as puppies got older.
Excretion
Reduced renal excretion, characteristic of the pediatric puppy, results in decreased clearance of renally excreted parent drugs and products of phase II drug metabolism. Although the number of glomeruli remains constant throughout pediatric development, both glomerular filtration and renal tubular function progressively increase.41,49 Adult values may not be reached until approximately 2.5 months of age. In contrast to glomerular filtration and secretion, renal tubular reabsorption in puppies appears to be similar to that in adults as long as body fluids and electrolytes are maintained.50,51 The sequelae of developmental changes in pediatric renal function include decreased clearance and prolonged half-life of drugs (primarily water soluble) excreted by the kidneys. Such a pattern has been shown for several drugs. Compared with current recommendations for adults, pediatric patients may require a higher dose (owing to increased volume of distribution) and longer intervals (owing to increased distribution and decreased clearance) for gentamicin administration. More important, modifications should be anticipated in the gentamicin dosing regimen of unhealthy puppies because they are likely to be affected by conditions that increase the potential for gentamicin-induced nephrotoxicity (e.g., dehydration). However, underdeveloped glomeruli may actually protect the pediatric patient from aminoglycoside-induced nephrotoxicity.21 Further investigations are needed to establish safe yet effective doses of gentamicin for the neonatal puppy or kitten.
Specific Drug Therapy for the Pediatric Patient
Fluid therapy
Pediatric patients are predisposed to dehydration because extracellular fluid is increased, renal capacity to conserve water is decreased, the ratio of surface area to body weight is large, and fluid loss through immature skin is greater.52 Fluids can be administered by several routes. Crystalloids administered rectally should be isotonic; rapid rectal absorption of hyperosmolar solutions can lead to life-threatening hyperosmolarity. Subcutaneous administration may be an acceptable route if small volumes of isotonic fluids are administered in patients with normal hydration. Intraosseous fluid administration is an acceptable route of administration if a central vein is not accessible.53 Oral rehydration is recommended as the preferred therapy for dehydration caused by diarrhea in human pediatric patients.54
Antimicrobial therapy
Beta-lactam antibiotics are generally the drugs of choice for pediatric patients whenever possible. Although drug half-lives are likely to be prolonged, they tend to be safe because they are characterized by a wide therapeutic index. Higher doses may be necessary to achieve desired peak PDCs because their distribution is greater. The time interval of administration can be prolonged to compensate for the longer half-life. Therapeutic drug monitoring should be used to improve the safety and efficacy of aminoglycosides whenever possible. Higher doses and longer intervals may be necessary to achieve recommended peak and trough concentrations. Amikacin, which is potentially less nephrotoxic (and more effective against Pseudomonas spp.) than gentamicin, might be preferred. Quinolones are very effective and, for most patients, safe antimicrobials. They are characterized by excellent tissue distribution. However, these drugs are not appropriate for large-breed pediatric animals because they can cause destructive lesions in the cartilage of long bones. Thus the author does not recommend these drugs as first choice for any pediatric patient. The use of disease-modifying agents containing glucosamine should be encouraged in any animal in which cartilage is growing (or repairing). The combination of a sulfonamide with trimethoprim or ormetroprim tends to be safe and effective for kittens and puppies. Oral tetracyclines should be avoided in nursing animals and before tooth extraction. Therapeutic indications for lincosamides and macrolides are limited for pediatric patients. Because both groups of drugs undergo extensive biliary secretion and enterohepatic circulation, they should not be used as first-choice antimicrobials. An exception should be made for Mycoplasma infections for which tylosin is the drug of choice. Metronidazole is the drug of choice for Giardia infections in dogs and cats, and it is often used for the treatment of anaerobic infections. Decreased clearance and prolonged half-life should be anticipated in kittens and puppies; lower doses and longer intervals may be necessary to prevent CNS toxicity. Enrofloxacin is one of the few drugs that has been studied well in neonatal to pediatric kittens.37 After oral administration, bioavailability of enrofloxacin (5 mg/kg) was at least 33% at 2 weeks of age, increasing to 50% at 4 and 70% at 6 and 8 weeks of age. Following intravenous and subcutaneous administration, the area under the curve compared to adult cats varied with the age of the kittens and route of administration. Neonates (younger than 2 weeks of age) presented as most “different,” with area under the curve at least twofold higher than that of any other age (4, 6, or 8 weeks), although elimination half-life was similar among the groups studied. In general, volume of distribution (Vd) was greater (up to twofold) at 4 and 6 weeks compared with the younger age groups and adults, indicating the need for a higher dose; because clearance was faster as well, elimination half-life was actually shorter than with adults.
Sedation, anesthesia, and analgesia
Opioid agonists are the preferred sedative, premedicant, or analgesic of some veterinary clinicians for pediatric patients.19 Although associated with marked cardiac and respiratory depression, the effects of opioid agonists are largely reversible with opioid antagonists. Bradycardia can be prevented in older pediatric patients by premedication with atropine or glycopyrrolate. Whereas the duration of fentanyl analgesia (nontransdermal patch) is generally too short to justify its use for adult patients, some clinicians prefer it for short-term intraoperative analgesia for pediatric patients because it minimally affects the cardiovascular system. Ketamine can be administered subcutaneously, intramuscularly, or intravenously for the restraint and immobilization of young cats. Response to ultrashort barbiturates such as thiopental and methohexital or similar agents (e.g., propofol) should be exaggerated in young animals because of decreased body fat and hepatic clearance. Dilution to a 1% to 2% solution is indicated to prevent over-administration.
Complications associated with intravenous administration can be reduced by rectal administration of either thiopental or methohexital in human pediatric patients. As a class the benzodiazepines can probably be used safely in pediatric patients. Elimination occurs primarily by hepatic metabolism and is likely to be slower in pediatric patients. Benzodiazepines are, however, characterized by a wide therapeutic index. Midazolam, the newest member of this group, is more potent, has a faster onset of action, and is more rapidly eliminated than diazepam. Although not approved for use in human pediatric patients, it has been used in this age group successfully to induce sedation.55 Inhalant anesthetics are preferred for maintenance anesthesia in veterinary pediatric patients. Halothane, methoxyflurane, enflurane, and isoflurane, and to a lesser degree, sevoflurane have been used. Hypotension is a complication of all gas anesthetics, however, and variable patient response necessitates close monitoring.
Pregnancy and Lacation
Maternal–Fetal–Placental Unit
Even the most simple pharmacokinetic representation of the maternal–fetal system is complex, being composed of at least three compartments: maternal, placental, and fetal. The pharmacokinetics of each compartment is determined, in turn, by its own rate of absorption, distribution, metabolism, and elimination.56–58 Pregnancy is further complicated by its dynamic nature, with dramatic changes in placental and fetal growth and in the physiology of the pregnant animal. All pharmacokinetic processes change in concert with the progression of pregnancy.
The placenta transfers nutrients and oxygen from the mother to the fetus and facilitates waste removal from the fetus. However, the placenta also has a number of metabolic functions, among them the synthesis of hormones, peptides, and steroids that are vital for a successful pregnancy. The placenta also presents a barrier to drug distribution from maternal blood into the fetus. Transporter proteins (e.g., P-glycoprotein [P-gp]) on both the maternal and fetal side influence drug movement; placental phase I and II drug-metabolizing enzymes also have been identified throughout gestation in the human placenta. Thus far, CYP1A1, 2E1, 3A4, 3A5, 3A7, and 4B1 and uridine diphosphate glucuronosyltransferases have been detected in the term human placenta. However, these barriers to drug movement from the mother to fetus presented by the placenta are not impenetrable. Extrapolation of data among species regarding maternal to fetal transfer of compounds is complicated by differences in placentation.59 Humans and rats have the least number of layers. However, the endotheliochorial placentation of carnivores differs from hemochorial placentation of humans only by the presence of maternal endothelium, and it is likely that this single fenestrated layer does not present a significantly greater barrier.60 The idea of absolute placental selectivity has been replaced with the realization that any drug administered to a pregnant animal might prudently be anticipated to cross the placenta, regardless of the degree of intimacy between fetal and placental membranes.56,57,61 The route of administration is also likely to determine the amount of placental transfer: Routes that result in higher plasma peak concentrations (i.e., intravenously, as an intravenous infusion, and in multiple doses) are likely to expose the fetus to higher drug concentrations. Further, a “depot phenomenon” described in the human placenta reflects the accumulation of selected lipid-soluble xenobiotics in the placenta, with the placenta possibly serving as a storage site.61
The primary mechanism of drug movement is passive diffusion, with the amount of transfer dependent on physiochemical properties; less commonly, active transporters and facilitated diffusion contribute to drug movement.61 Although many factors determine the rate and extent of drug transfer across the placenta, the lipid solubility of the drug and a steep maternal–fetal drug concentration gradient are probably the most important.57 In general, nonionized compounds with high lipid solubility cross rapidly, whereas drugs with little lipid solubility cross slowly. Impermeability of the placenta to polar compounds, which generally do not penetrate cell membranes, has been described as relative rather than absolute.57 A number of drugs that are polar at physiologic pH can cross the placenta rapidly.57 Molecular weight influences drug movement, with those below 500 Dalton more likely and those above 1000 less likely to transfer. Protein binding precludes drug movement, whereas differences in fetal and maternal blood pH increase the accumulation of weak bases in fetal blood (humans).
Fetus and Neonate
Unique differences in drug disposition predispose the near-term fetus and neonate to adverse drug reactions. The pharmacologic principles that address risk of fetal exposure to potential teratogens have been reviewed.61,62 The drug approval process generally precludes availability of those compounds with a demonstrated risk, but premarket assessment does not necessarily occur in dogs or cats. The responses of the fetus and newborn to individual drugs, however, vary. Differences in responses reflect, in part, differences in placental kinetics of drugs. The fetus is most sensitive to adverse effects particularly in the first trimester. The risk is positively correlated with the duration of pregnancy, with the risk greater in shorter pregnancies (e.g., dogs and cats) because this period represents a longer proportion of the pregnancy.63
Current efforts in human neonatology are concerned with the characterization of the pharmacokinetic differences between drugs in the near-term fetus. Differences in drug disposition compared with that of both pediatric animals and adults can lead to adverse reactions in the near-term fetus receiving drugs through the placenta. The amount of fetal protein is generally less in the neonate, which is, in turn, less than that in the adult.56,57 Thus higher concentrations of unbound and pharmacologically active drugs can be anticipated. It is not clear if increased concentrations of unbound drug will be compensated for by increased clearance, as will occur in the adult animal. Perhaps more important are anatomic peculiarities of fetal circulation. Because the fetal liver and lungs are largely bypassed, blood reaching the heart and brain contain essentially the same concentration of drugs as present in the umbilical vein. Although fetal metabolism of drugs can contribute to the ultimate elimination of drugs in the human neonate, the amount of drug-metabolizing enzymes present in near-term animals is negligible.57
Although drugs administered to pregnant animals may be detectable in the fetus, they may not produce clinically important effects. Examples of drugs that have been shown to reach detectable and potentially clinically important concentrations in the fetus include salicylates and other NSAIDs, anticonvulsants (phenytoin and diazepam), local anesthetics such as lidocaine, gentamicin (in some species), and narcotic analgesics. In human infants the ratio of maternal to fetal concentration of beta-lactams approximates 1.64 Because predicting the effects of a drug crossing the placenta is difficult, drug selection for the mother should be based, in part, on anticipated safety to the near-term fetus.
Maternal
The effects of pregnancy can alter all phases of disposition in the mother. Gastrointestinal motility and gastric acid secretion decrease and may lead to decreased drug absorption. Distribution may be influenced by decreased serum albumin, and increased Vd of drugs, resulting in lower PDCs. However, drug clearance may be more rapid as cardiac output, renal blood flow, and glomerular filtration rate increase. High progesterone concentrations may induce hepatic microsomal enzymes and increase drug metabolism.65
Lactation
As is the fetus, the nursing animal is an inadvertent recipient of drugs administered to the mother. Most of the pertinent information in the veterinary literature is concerned with excretion of drugs in the milk of food animals; there appears to be no information regarding small animals. Studies of humans indicate that drugs diffuse into the milk from maternal circulation. Low-molecular-weight (<200), un-ionized, highly lipid-soluble drugs that are minimally protein bound diffuse into the lactating mammary gland rapidly, whereas water-soluble drugs diffuse more slowly.66 The pKa of a drug largely determines its concentration in milk. Animal milk tends to be acidic compared with plasma pH. Consequently, although a drug may be nonionized in the plasma and thus more likely to diffuse into milk, it may become ionized and nondiffusible once in the milk. Such “ion trapping” can concentrate drugs in milk. The ratio of drugs in milk to plasma is predictable, being greater for weak bases and weak acids whose pKas differ from the pH of milk by 2 pH units (+2 for acids and −2 for bases).67 Generally, the amount of drugs excreted in milk is less than 2% of the maternal dose.66 Greater concentrations can be expected, however, if a drug is administered to the mother intravenously, as an intravenous infusion, or in multiple doses.
Not all drugs ingested with milk during nursing will be absorbed from the gastrointestinal tract of the nursing animal. For example, milk may decrease the absorption of some drugs, whereas the pharmacokinetic properties of other drugs (i.e., aminoglycosides) preclude their absorption except in the very young. Not all drugs, however, must be absorbed to cause clinically important adverse effects. For example, antimicrobials can sometimes alter the developing flora of the pediatric alimentary tract.27,68 Thus it is prudent to refrain from administering potentially toxic drugs to the lactating bitch or queen.
Sex
Pregnancy and lactation are obvious differences between the sexes that affect the disposition of and response to drugs. However, sex differences are relevant without the influence of active reproduction. The impact of sex on drug disposition is not well characterized in human or other animal medicine. In general, female humans, compared with male humans, are characterized by reduced smooth muscle motility, greater body fat, greater content and fluctuation in plasma volume and increased organ blood flow, xenobiotic metabolism (increased CYP3A4, others not clear), and transporter protein activity.69,70 It should be anticipated that sex differences are likely to be complex, as has been demonstrated for CYP enzymes in cats.71 Female cats were characterized by greater CYP2D and lower CYP3A compared with males.
Roles of Species and Breed Differences in Drug Disposition
Differences among species in the kinetics and response to drugs are profound; Riviere72 has offered a review in animals. Few studies have compared disposition among species, and when reported, data are generally oriented toward human medicine, with the intent to identify the (laboratory) animal most predictive of human drug disposition. Presumably, species that are physiologically similar tend to have similar drug disposition patterns, and the same dosing regimen can often be used for a particular drug.73 However, pharmacokinetic data (Vd, clearance, and mean residence time) after intravenous administration have been compared among humans, monkeys, dogs, and rats for over 100 molecules (including many drugs).74 Correlations between chemical structures of the compounds with pharmacokinetics allowed two categories of compounds to be identified: those characterized by high clearance, for which mathematical models would allow general extrapolation of data from one species to another, and those whose characteristics yielded incorrect extrapolations. Although the authors concluded that such an approach might be useful for extrapolation, more to the point is the reality that drugs may not be extrapolatable even with the most sophisticated of mathematical models. The combination of computation analysis of the molecular characteristics of each compound with in vivo pharmacokinetic data might ultimately be helpful in identifying the most appropriate species for predicting pharmacokinetic behavior of a particular molecule in another species. Thus caution needs to continue when extrapolating information from humans to the dog, and dog to cat. Likewise, pharmacodynamic responses may be profound.
Absorption
Gastric pH profiles have been described for the dog in anticipation of extrapolation to humans.75 Dogs are described as poor gastric acid secretors, compared with humans as good secretors, with gastric pH fluctuating in dogs from 2.7 to 8.3 (mean 6.8). The shorter gastrointestinal tract of dogs compared with that of humans, results in a transit time (111 minutes) that is 50% of that of humans, although this may be offset by taller villi and greater bile salt concentrations. Accordingly, differences in absorption might be expected between dogs and humans in the oral absorption of drugs, particularly for enteric-coated or altered-release products.75 Different affinities for P-gps have been demonstrated among humans, nonhuman primates, and Beagle dogs.76 In dogs absorption of some hydrophilic compounds is more similar to that in rats, whereas absorption of others is more similar to that found in humans.77 Another retrospective comparison between dogs and humans in the oral bioavailability of 43 drugs found close to 50% to be completely absorbed in both species. For the remaining drugs, 12 were absorbed more rapidly and to a greater (15% to 200%) extent.78 However, the extent of absorption for drugs not well absorbed correlated poorly between the two species. In general, if the drug was well absorbed in humans, it tended to be well absorbed in dogs. Both lipid- and water-soluble drugs appear to be better absorbed in dogs, the latter suggesting more paracellular transport. The rate and magnitude of drug absorption for many drugs may be similar between dogs and cats, regardless of the route of administration. However, extrapolations must be done with caution, as is exemplified with ciprofloxacin, whose oral bioavailability in the dog is 40% (compared to 80% or better in humans) but 0% to 20% (the latter more likely with multiple dosing) in cats.79,80 Another example is prednisone. An exception also may need to be made for slow-release preparations; rates and extent of absorption do vary among species. Dye81,82 has demonstrated not only differences in bioavailability in cats versus dogs but also differences in pharmacokinetics related to morning versus evening dosing. Because slow-release preparations used in human patients are designed to maintain therapeutic concentrations in humans, absorption kinetics of these products can be profoundly different in the dog and cat. Use of such drugs should be based on clinical studies of these preparations in dogs and cats.
Distribution
Because most determinants of xenobiotic distribution are largely affected by cardiac output, regional blood flow, and xenobiotic chemistry, distribution differences among species might be predictable through allometric scaling.83 However, they must also take into account differences in transport proteins. Differences in drug distribution can result in important differences in drug response. Blood volume of the cat (70 mL/kg) is less than that of the dog (90 mL/kg); PDCs of drugs whose distribution is confined to the plasma compartment may therefore differ between the species. The same amount of drug (on a per-kilogram basis) is diluted less in cats because the plasma volume is smaller. Thus drug concentrations after administration of a mg/kg dose might initially be higher in cats than in dogs. Organs that are well perfused (i.e., heart, brain) may be more susceptible to toxicity. Cats are approximately the same size as the smaller dog breeds. Thus doses determined for medium-size to large-size dogs may not be appropriate for the cat because the smaller animals have a greater body surface area. In larger animals body water makes up a larger proportion of body weight, which tends to dilute the drug. A higher dose may be needed for larger animals. Because the drug half-life may be longer (owing to increased distribution), however, the dosing interval may need to be longer.
Differences in plasma protein-binding characteristics (particularly albumin) may alter the distribution of drugs that are highly protein bound. The degree to which various drugs are protein bound varies dramatically among the species, although the clinical implication is not clear. Although the elimination characteristics of many drugs have been established for cats, few studies have determined the extent of protein binding.
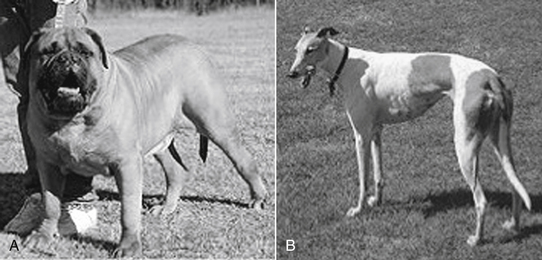
Sight hounds (e.g., Salukis, Greyhounds; Figure 2-3) offer an example of potential breed differences in drug distribution. Their lean body weight provides little fat tissue for drug distribution. As a result, they are more susceptible to overdosing with drugs that redistribute, such as thiobarbiturates.
Distribution is influenced by the multidrug transport protein P-gp. Polymorphism has been demonstrated in the Collie, yielding differences in P-gp content. Two P-gp transporting proteins are encoded by ABCB1 (previously MDR1 or PGY1) and MDR3 (also named MDR2 and PGY3); only the MDR1 gene product is thought to significantly influence drug metabolism. P-gp acts as an efflux pump by translocating drugs from the intracellular to extracellular compartments (Table 2-2) (see Chapter 3). The protein transports a large number of drugs that are chemically divergent; further, these drugs are associated with a specific CYP450 responsible for metabolism of the drugs that have been transported. Polymorphism of the MDR1 gene and P-gp have been reported in humans and are associated with altered drug disposition and thus susceptibility to adverse drug events. Interestingly, polymorphism also has been associated with an increased risk of certain illnesses (e.g., refractory seizures, Parkinson’s disease, inflammatory bowel disease biliary mucoceole in dogs∗). Polymorphism reflecting a mutation deletion of MDR1 that causes nonfunctional P-gp has been documented in Collie and related working-breed dogs. The incidence of the deletion is impressively high: in the U.S., in one study, 35% of Collies were homozygous and another 42% heterozygous for the mutation deletion.84 A similarly high incidence was found in dogs in France: 20% of Collies and related breeds were found to be homozygous for the normal allele, 32% heterozygous for the deletion (carrier), and 48% homozygous for the mutant allele (affected dogs).85 The impact of the mutation on drug safety in afflicted animals can be profound (see Chapter 3). Substrate specificity for P-pg appears to be similar among species, suggesting that human data can be used to predict which drugs might be more likely to cause adverse effects in these breeds.86 However, protein type and amount may vary among species and breed.
Table 2-2 Substrates for P-glycoprotein Transport Pump and Known Inhibitors and Inducers of Drug Transport
| Substrates | Inhibitors | Inducers |
|---|---|---|
| Antimicrobial Drugs | ||
| Erythromycin | Bromocriptine | Clotrimazole |
| Tetracycline | Carvedilol | Dexamethasone |
| Itraconazole | Cyclosporine | Morphine |
| Fluorinated quinolones (selected) | Erythromycin | Rifampin |
| Anticancer Drugs | Fluoxetine | Phenothiazine |
| Doxorubicin | Intraconazole | St. John’s Wort |
| Vinblastine | Ketaconazole | |
| Vincristine | Meperidine | |
| Mitoxantrone | Pentazocine | |
| Anthelmintics | Progesterone | |
| Ivermectin | Quinidine | |
| Cardioactive Drugs | Tacrolimus | |
| Digoxin | Verapamil | |
| Quinidine | ||
| Diltiazem | ||
| Verapamil | ||
| CNS-Active Drugs | ||
| Phenothiazines | ||
| Amitryptyline | Inhibitor of CYP3a | |
| Morphine | Substrate CYP3a | |
| Endogenous Substrates | ||
| Bilirubin | ||
| Steroidal Hormones | ||
| Cortisol | ||
| Aldosterone | ||
| Gastrointestinal Drugs | ||
| Cimetidine | ||
| Loperamide | ||
| Ondansetron | ||
| Tacrolimus | ||
| Immunomodulators | ||
| Dexamethasone | ||
| Methylprednisolone | ||
| Cyclosporine | ||
| Tacrolimus | ||
| Colchicine | ||
CNS, Central nervous system.
Metabolism
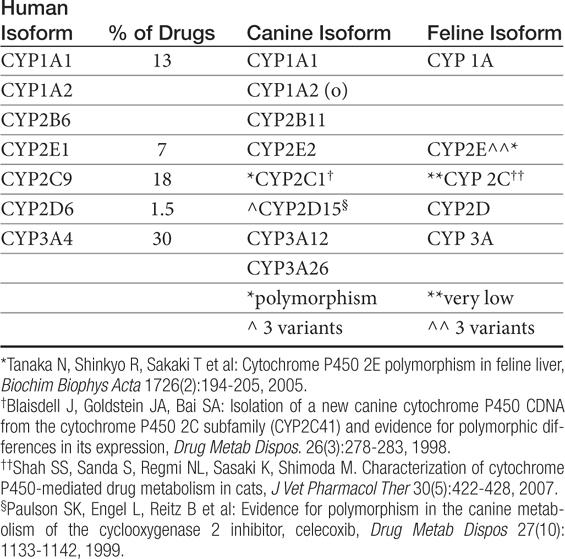
Human polymorphisms in CYP metabolic enzymes have been associated with therapeutic failure resulting from extremely rapid metabolism of a drug and toxic effects caused by decreased metabolism.87 Species extrapolation among the CYP450 appears to be predictable in order of most to least: 2E1 (reasonably predictable) greater than 1A1, 1A2, and 4A (cautiously predictable) greater than 2D and 3A greater than 2A, 2B, and 2C (major caution with extrapolation).88 Studies attempting to identify similarities between dog and human CYP activity suggest general extrapolation between the species is not prudent.87 Although significant differences among species were not detected among rodents, rabbits, and dogs for CYP2E1, 3A2, and 4A1, comparison of animal CYP activity with human found only 2D6 to be most similar to dog.89 Other enzymes present in dogs are CYP1A, 2C, and 2D families and subfamilies (Table 2-3).90
Table 2-3 Cytochrome P450 Families, Their Contribution to Drug Metabolism in Humans, and Their Orthologs in Other Species
Species differences have been documented in the handling of racemic isomers of selected drugs. Inversion (from one isomer to the other) patterns differ and are likely to result in different pharmacologic or toxic effects of the drugs. Induction (or presumably inhibition) of drug-metabolizing enzymes also will differentially affect the isomers.91
As in humans, polymorphism in drug-metabolizing enzymes has been reported in dogs92 but is not as well described. Differences in response to anesthesia recognized in sight hounds reflect both differences in drug distribution (to lean versus fat compartments; see Chapter 1) as well as differences in metabolism. Cytochrome-mediated clearance of several anesthetic agents is less in Greyhounds compared with other (nonsight hound) dogs; documented drugs include thiopental, thiamylal, and methohexital. Clearance of propofol by Greyhounds is three times less than that by Beagles. Ketoconazole plasma concentrations were twofold higher than expected in Greyhounds in one study.93 Further, Greyhound disposition of celecoxib, a cyclooxygenase-1 protective NSAID, indicates that breed differences may predispose this breed to adverse drug reactions. Polymorphism also has been described for CYP2C isoenzymes, again in Beagles94 and possibly Greyhounds.95 Polymorphism in celecoxib metabolism was attributed to CYP2D15, for which three canine variants were found.96 In a study of 242 Beagles receiving celecoxib, approximately 50% were considered efficient metabolizers and 50% poor metabolizers, with bioavailability and maximum PDC in the latter group almost twofold higher. The impact of species differences in pharmacokinetic and pharmacodynamic considerations of enantiomers has been described.91,97 For example, for many NSAIDs the S-isomer has a much greater affinity for cyclooxygenase-2, but the proportion of the S isomer varies among species. Further, species differ in their ability to interconvert S and R isomers.97 For example, chiral inversion has been described for ketoprofen in cats.98Other polymorphisms that have been described include thiopurine methyltransferase (TPMT), which is one of several enzymes responsible for the metabolism of the active metabolite of the prodrug azathioprine; polymorphisms resulting in deficiencies in humans have been associated with an increase in the toxic bone marrow effects of the drug. Differences in dogs have been demonstrated as well: Kidd99 and coworkers have demonstrated that Giant Schnauzers have significantly less and Alaskan Malamutes significantly more TPMT compared with other canine breeds.
The most significant and best-characterized differences in drug disposition between the dog and cat probably result from differences in drug metabolism. Identification of phase I enzymes and their specific drug substrates is difficult, and few species differences have been described in the cat or dog. However, a recent review of CYP450 activity–based substrate metabolism in cats suggests that cats have very low activity of CYP2C, but activity of CYP2D and CYP3A approximates that of dogs or humans, depending on sex.77 Deficiencies in demethylation and hydroxylation have been described in the cat and may be responsible for different patterns of prodrug activation (e.g., primidone; see Chapter 27) or adverse reactions to selected drugs (i.e., chloramphenicol).100 Deficiencies in phase I demethylation and hydroxylation as well as phase II glucuronidation lead to much slower elimination of phenols and aromatic acids and amines in the cat compared with other species (Figure 2-4).101,102 The described reaction of cats to diazepam may represent differences in the metabolites produced, as may the susceptibility of the feline liver to metabolite-induced damage.103 Polymorphisms in drug-metabolizing enzymes have also been described in cats, which have at least three variants of CYP2E,104, although breeds were not cited.104
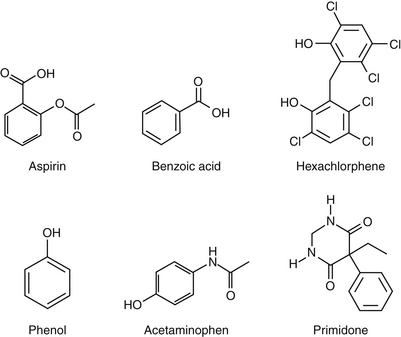
Deficiencies in phase II metabolism have long been recognized in cats. The deficiency reflects extremely low concentrations of some glucuronyl transferases. Thus many drugs excreted as glucuronide conjugates in other species are characterized by a prolonged clearance rate and half-life in the cat. Toxic levels may accumulate much more quickly in the cat, and exaggerated pharmacologic responses or toxicities occur more easily (Figure 2-4). Dosing regimens must be modified for such drugs by either decreasing the dose (especially for drugs whose dosing interval is shorter than the elimination half-life) or prolonging the dosing interval. The prototypic example is aspirin, whose half-life approximates 36 hours in cats compared with 8 hours in dogs. To prevent toxicity in the cat, aspirin is dosed every 48 to 72 hours, compared with twice daily in dogs.
Not all drugs that are conjugated with glucuronide are predisposed to toxicity in the cat. This is true for several reasons. First, the cat is deficient only in certain families of glucuronyl transferase. Cats can conjugate and excrete endogenous substrates such as bilirubin, thyroxine, and steroid hormones as well as other species. Metabolism of a variety of exogenous drugs, however, particularly phenols and aromatic acids and amines, occurs at a much slower rate in the cat than in other species.101,102 The degrees of deficiency and potential toxicity depend on the drug substrate. For example, some phenolic compounds are sufficiently conjugated, whereas others are not. Second, glucuronide-conjugated drugs characterized by a wide safety margin are associated with few adverse reactions even if accumulation occurs. Finally, in the absence of glucuronide, drugs may be sufficiently metabolized by an alternative pathway. Some sulfates may be particularly well developed in the cat, and many drugs that are excreted as glucuronide-conjugates by the dog may be excreted as sulfated compounds by the cat. Other sulfate-conjugating systems, however, appear to be easily saturated in the cat. Unfortunately, alternate pathways of drug metabolism may also contribute to the toxicity of some drugs because they may involve phase I enzymes that catalyze the formation of toxic metabolites. Thus drugs shunted to another pathway in the cat may be very toxic to the cat but minimally toxic in other species. Deficiencies in both glucuronide and (potentially) glutathione transferase may also predispose the cat to poor scavenging systems in erythrocytes and hepatocytes, limiting the otherwise protective effects that might be realized by these systems and further contributing to toxicity. Acetaminophen is an excellent example of the potential sequelae of phase II deficiences in the cat (See Chapter 4). Because glucuronide is deficient, excessive acetaminophen is shunted to phase I enzymes, which produce toxic oxygen radicals. More metabolites are produced than can be handled, and the glutathione-scavenging system of feline erythrocytes and hepatocytes is overwhelmed, resulting in life-threatening methemoglobinemia and (potentially) hepatic necrosis. The rationale for cimetidine treatment can be understood in the context of the role of phase I metabolism, as well as that of N-acetylcysteine, a glutathione precursor.
Not all deficiences in metabolism occur in the cat. Although acetylation is not a common route of elimination for xenobiotics, it is an enzyme system whose deficiency in the dog is clinically relevant (e.g., procainamide). For example, the antiarrhythmic procainamide is acetylated in humans to an active metabolite. Procainamide is less potent than its acetylated metabolite and the canine dose for procainamide is considerably higher than that in humans on a mg/kg basis in order to achieve an equivalent pharmacologic response (see Chapter 14, Cardiac). A second example might be sulfonamide elimination. Sulfonamides are detoxified by N-acetylation in humans. In the face of deficient acetylation, shunting of the xenobiotic to an alternative pathway in dogs, with the production of the cytotoxic metabolite hydroxylamine, may be one mechanism of sulfonamide toxicity in dogs, although alternative mechanisms are likely to be responsible.105
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


