Fig. 1.1.
Eyeblink conditioning paradigms typically used in human and rabbit studies of Alzheimer’s disease and in rat studies of fetal alcohol spectrum disorders. In the 400-ms delay paradigm (1), a 500-ms conditioned stimulus (CS) precedes, overlaps, and coterminates with a 100-ms airpuff unconditioned stimulus (US). In the 750-ms delay paradigm (2), an 850-ms CS precedes, overlaps, and coterminates with a 100-ms airpuff US. In the 750-ms trace paradigm (3), a 250-ms CS precedes a 100-ms airpuff US. A 500-ms stimulus-free “trace” interval is imposed between the offset of the CS and the onset of the US. The 750-ms delay and trace paradigms are matched for the 750-ms interstimulus interval (ISI) between CS and US onset. In the 280-ms delay paradigm often used in studies with rats (4), a 380-ms CS precedes, overlaps, and coterminates with a 100-ms periocular shock US. The 880-ms delay paradigm (5) is also typically used in rat studies and consists of a 980-ms CS that precedes, overlaps, and coterminates with a periocular shock US. Parameters for auditory CSs used in these studies range between 1–2.8 kHz and 70–85 dB.
1.1 Fetal Alcohol Syndrome
In addition to the neurobiological parallels among mammalian species, there are dramatic parallels in eyeblink conditioning in development over the life span. Most of the life-span data have been collected in adulthood, where it has been demonstrated that age-related eyeblink conditioning deficits appear in rats, rabbits, and humans in middle age. A basis for drawing parallels between rodent and human developmental models was demonstrated with the description of techniques and results with eyeblink classical conditioning in 4- and 5-month-old infants (8). This approach has the potential to offer fundamentally important data on the early development of cognitive and neurobehavioral disorders.
1.1.1 Human Studies of Fetal Alcohol Spectrum Disorders
Prenatal alcohol exposure is associated with a variety of physical, behavioral, and neuropsychological impairments and in some cases, fetal death. Fetal alcohol syndrome (FAS), the leading preventable cause of mental retardation in the Western world (9), is characterized by a distinct facial appearance (indistinct philtrum; thin upper lip; short palpebral fissures), CNS dysfunction (microcephaly; IQ deficits), and growth impairments (at or below tenth percentile for height or weight) resulting from heavy prenatal alcohol exposure in humans (10–14).
Fetal alcohol spectrum disorders (FASDs) is a term used to encompass the range of effects resulting from prenatal alcohol exposure (15, 16) and is associated with widespread brain damage and impairments in many domains, including executive functioning/attention, motor coordination, spatial memory, and social functioning (13, 17–21). Incidence of FASDs in the USA is estimated at 0.9–1 of every 100 births (14, 22), with annual costs exceeding $4 billion (23).
1.1.1.1 Anatomical Abnormalities in Fetal Alcohol Spectrum Disorders
Certain brain regions appear to be particularly susceptible to prenatal alcohol exposure, including the corpus callosum, basal ganglia, specific cerebral cortical regions, and – most relevant to this review – the cerebellum (13, 24–26). The cerebellum is associated with motor coordination, including balance, postural adjustment, and associative learning (e.g., eyeblink conditioning). Cerebellar dysmorphology in the form of region-specific hypoplasia and displacement is among the most widely reported anatomical abnormalities in the FASD literature (24, 27–31). Impairments in domains of motor functioning (i.e., balance; coordination) have been cited as well (12, 32, 33). Roebuck et al. (34) examined balance and postural sway in FASD children and controls to assess possible cerebellar-related impairments. The Sensory Organization Test was used to assess the influence of various modes of sensory information on the maintenance of balance. When visual and particularly somatosensory information was compromised or absent, FASD children performed more poorly (as measured by postural stability). However, no group differences emerged when only visual information was compromised, suggesting that cerebellar dysfunction may make FASD children particularly sensitive to altered somatosensory input.
These findings are significant in that they demonstrate the importance of task selection in identifying alcohol-induced impairments. Furthermore, while the deficits appear to be cerebellar dependent, possible effects on peripheral mechanisms cannot be ruled out. To obviate such potential confounds, cerebellar-dependent tasks that limit demands on multi-joint coordination may be ideal (c.f., 35). Toward that end, eyeblink classical conditioning may provide clearer indications of cerebellar dysfunction in FASDs (further details on eyeblink conditioning in the study of FASDs are presented below).
1.1.2 Rat Models of Fetal Alcohol Spectrum Disorders
Various animal models involving early developmental alcohol exposure have produced CNS deficits similar to those observed in human FASD. The period of most rapid brain growth that is often most susceptible to environmental insult (termed the “brain growth spurt”) occurs at different relative points in ontogeny across species and must be taken into consideration (36). In rats, the brain growth spurt occurs within the first 2 weeks of postnatal life. This period of brain development in rats is considered “developmentally equivalent” (e.g., in terms of region-specific synaptogenesis and other critical developmental stages) to third trimester human fetuses. The rat model of FASD is useful in that (a) neurological anomalies (e.g., cerebellar damage) in rats exposed to alcohol during critical developmental periods are similar to those observed in human FASD, and (b) because of the extensively documented behavioral capacities of rats, demonstrating functional (behavioral) impairments resulting from early alcohol exposure is easier to demonstrate relative to other species.
The rodent cerebellum, like in humans, is highly sensitive to early developmental alcohol exposure. Studies of alcohol-related cerebellar damage typically focus on Purkinje cells, prenatally generated neurons that receive massive sensory input and represent the sole output of the cerebellar cortex. Early studies revealed Purkinje cell depletion following alcohol exposure spanning prenatal and postnatal periods (GD12–PD4; 37). While alcohol exposure (blood alcohol concentrations (BACs) > 400 mg/dl) restricted to gestation is capable of depleting Purkinje cell populations (38), the neonatal period (“brain growth spurt”) appears to be a particularly sensitive period. Marcussen et al. (39) found reduced vermal Purkinje cell numbers at PD10 in subjects exposed postnatally (PD4–9; BACs ∼ 200 mg/dl) but not in those exposed prenatally (GD13–18; BACs > 250 mg/dl). Enhanced sensitivity during the neonatal period was further demonstrated by Maier et al. (40), as exposure during all three trimesters equivalent (gestation plus PD4–9) or the “third trimester equivalent” only reduced Purkinje cell populations, with no such impairments observed following alcohol exposure limited to gestation.
1.1.2.1 Binge-Like Exposure Models
Early work was successful in achieving substantial cerebellar microcephaly (small brain relative to body size) and reductions in cerebellar neuronal populations (Purkinje cells; granule cells) at PD10 following postnatal (PD4–10) binge-like exposure to high alcohol doses (BACs > 450 mg/dl; 41). Later studies employing alcohol delivery over this period demonstrated that (a) more moderate doses (4.5 g/kg) are sufficient to produce brain damage, (b) the pattern of exposure is critical, as lower doses (4.5 g/kg) condensed into more frequent deliveries (binge-like) produce greater cerebellar damage than a higher dose (but lower BACs; 6.6 g/kg) gradually delivered over a longer period, and (c) brain damage persists into adulthood (42, 43).
Alcohol was delivered via artificial rearing in some of the early studies (e.g., 42, 43). Artificial rearing entails surgical implantation of feeding tubes into the stomach which justifiably raises concerns over unwanted surgery/stress effects. Another delivery method is exposure to ethanol vapors, which results in comparable BACs and Purkinje cell depletions (44, 45). Both delivery methods include extended periods of maternal separation. Intragastric intubation, a method in which a polyethylene tube is inserted down the esophagus and into the stomach for alcohol delivery (46, 47), avoids these concerns as maternal separation is minimal.
1.1.2.2 Regional Selectivity and Critical Periods in the Neonate
An outcome of neonatal alcohol exposure that is robust across a variety of doses, patterns of dosing, and delivery methods are findings of enhanced regional vulnerability. Deficits are consistently shown in earlier-maturing (i.e., I, II, XI, X) relative to later-maturing cerebellar cortical lobules (i.e., VI, VII; 4, 42, 43, 45, 48). Furthermore, neonatal binge-like alcohol effects upon Purkinje cells depend both on dose and exposure period within the PD4–9 window (the PD4–9 dosing period has often been used in “third trimester” equivalent models). While daily neonatal alcohol exposure to 4.5 g/kg+ (BACs generally > 200 mg/dl) is typically used to obtain Purkinje cell depletion (e.g., 42, 46, 49), slightly lower doses delivered in a binge fashion (i.e., 3.3 g/kg delivered across two feedings or 2.5 g/kg at once – BACs ∼ 150–200 mg/dl) are also capable of producing moderate neuronal loss (39, 50).
1.1.2.3 Behavioral Effects
Alcohol-induced brain damage is truly problematic insofar as it compromises quality of life, vis-à-vis behavioral and cognitive impairments. The primary behavioral deficits reported as a result of neonatal exposure in rodents are generally in the spatial and motor domains. Neonatal alcohol exposure also impairs traditionally recognized cerebellar-dependent tasks that place high demands on motor control (i.e., parallel bar and rotating rod; 51–53). Though Purkinje neuronal loss has been correlated with motor performance deficits (53), establishing links between alcohol-induced cerebellar damage and behavior is difficult in these instrumental tasks that require complex multi-joint coordination. Cerebellar-dependent Pavlovian tasks like eyeblink classical conditioning which utilize an easily measurable reflexive response and engage a well-defined neural circuit (54) provide a more promising avenue for investigation of causal relationships between alcohol-induced brain damage and behavior (35, 55).
1.1.2.4 Eyeblink Conditioning in Rats
The extensive behavioral (56) and neurobiological (54) data compiled for the first two decades of research on animal models of eyeblink conditioning has focused on the rabbit model, due in part to their tolerance for restraint. Because rats do not accept restraint as well as rabbits, their testing in eyeblink conditioning requires a freely moving preparation. This was established by Ronald Skelton in the late 1980 s using adult Long Evans rats (57). Mark Stanton adapted this freely moving preparation to developing rats. Specifically, the seminal report on this body of work (58) showed that eyeblink conditioning in the rat using a short-delay paradigm (280 ms interstimulus interval – ISI – between CS and US onset – Fig. 1.1) develops gradually between postnatal days (PD) 17 and 24. Furthermore, acquisition differences across ages cannot be attributed to differences in CS or US processing (58), increasing US intensity or varying the ISI does not overcome failures of conditioning at PD17 (59), and the learning (conditioned eyeblink responses (CRs)) present in weanlings is cerebellar dependent (60, 61). John Freeman and colleagues have replicated the behavioral emergence of eyeblink conditioning in rats (PD17–24; see 62) and, through electrophysiological recording of various components of the putative cerebellar–brainstem “eyeblink” circuit, have offered insights into the underlying mechanisms of the ontogeny of eyeblink conditioning.
1.1.3 Fetal Alcohol Spectrum Disorders Models and Eyeblink Conditioning
The neonatal binge alcohol exposure paradigm results in damage to multiple components of the critical eyeblink conditioning circuit (42, 63, 64). In an effort to translate neurological impairments suffered as a result of neonatal alcohol exposure as directly as possible to functional (behavioral) impairments in cerebellar-mediated tasks, Mark Stanton and Charles Goodlett adapted the neonatal binge alcohol rat model to the freely moving developing rat eyeblink preparation (65). Using the artificial rearing method of binge-like alcohol delivery at doses of 5.25 g/kg/day to Long Evans rats over PD4–9, Stanton and Goodlett (65) reported significant impairments in conditioning in alcohol-exposed rats relative to controls when eyeblink testing occurred over PD23–24. Importantly, these impairments appeared to be associative in nature, as CS and US processing were unimpaired as a result of neonatal alcohol exposure.
Subsequent reports applying the neonatal binge exposure model (5 g/kg/day of alcohol) to the freely moving rat eyeblink preparation have utilized the intragastric intubation method and have advanced the findings of Stanton and Goodlett (65) by (a) further showing that impairments in conditioned responding appear to be associative in nature, as unpaired CS–US training does not yield robust CR generation differences as a function of neonatal treatment (66, 67), (b) associative deficits persist into adulthood (66, 68–71), and (c) CR deficits are correlated with cerebellar neuronal loss (67, 69) and impaired cerebellar electrophysiological activity (68). This rodent model appears to be applicable to the human condition, as children with FAS are also impaired in acquisition of eyeblink classical conditioning (72, 73). Furthermore, recent studies from Mark Stanton’s laboratory have revealed deficits in eyeblink conditioned responding in both post-weanling (74) and adult rats (75) following PD4–9 binge exposure to alcohol doses as low as 3–4 g/kg/day. These dose-dependent deficits were revealed when using more complex variants of eyeblink conditioning – e.g., ISI discrimination – relative to the single-cue paradigms typically employed. In ISI discrimination, two distinct CSs (tone and light) are paired with the US at two different CS–US intervals (280 and 880 ms ISI – Fig. 1.1).
1.2 Alzheimer’s Disease
Severe memory loss is the most prominent clinical symptom of AD, and this memory impairment has long been associated with impairment in acetylcholine neurotransmission. Disrupted cholinergic neurotransmission is not the single cause of memory loss in AD, but this deficit characteristic of AD clearly impairs memory (76). Research in our laboratory testing patients diagnosed with AD began with an idea based on results from the rabbit eyeblink conditioning experiments. We knew from the animal studies that acetylcholine neurotransmission in the medial temporal lobes (specifically, the pathway from the medial septum into the hippocampus) was activated during eyeblink conditioning. Based on the results with the animal model, we hypothesized that patients with a diagnosis of probable AD would be impaired in eyeblink conditioning beyond the deficit due to normal aging, because AD damages the cholinergic system of the human brain (77).
Our hypothesis that eyeblink conditioning would be severely impaired in patients with AD was initially confirmed in a sample of 40 elderly adults, half of whom were diagnosed with probable AD (78). There were very significant differences in eyeblink conditioning between the patients and age-matched, non-demented control subjects. Normal older adults are impaired in eyeblink classical conditioning compared to young adults, but all age groups of normal, non-demented adults, including adults in their 80 s and 90 s, show clear evidence of associative learning (79). In probable AD, there is very limited eyeblink conditioning in the first session of testing. However, when given a sufficient number of training trials (e.g., 4 or 5 days of 90-trial presentations), patients diagnosed with probable AD acquire conditioned eyeblink responses (called CRs; 80, 81). This slowing of the rate of acquisition occurs in the animal model when antagonists to acetylcholine neurotransmission are introduced (82, 83). There is strong evidence that the site of interference of cholinergic antagonists in rabbits is the hippocampus (84). The facts that patients with AD are slow to acquire CRs and that AD patients do eventually acquire CRs are parallel to results in rabbits injected with antagonists to acetylcholine. Disruption of the hippocampal cholinergic system prolongs acquisition of CRs, but organisms eventually condition.
Solomon et al. (85) reported disruption of eyeblink conditioning in the 400-ms delay paradigm (Fig. 1.1) in a sample of probable AD patients in their early 70 s – a decade younger than the Woodruff-Pak et al. (78) sample. Woodruff-Pak et al. (86) carried out an additional replication in the 400-ms delay procedure testing 28 patients with probable AD and 28 healthy age-matched control subjects. Patients diagnosed with cerebrovascular dementia were also tested for this study, and eyeblink conditioning was shown to differentiate some cerebrovascular dementia patients from patients with probable AD. Eyeblink conditioning in adults over the age of 35 years with Down’s syndrome and presumably AD was similar to conditioning in probable AD patients (87, 88). Conditioning in patients with neurodegenerative disease that does not affect the critical cerebellar eyeblink conditioning circuitry or disrupt the hippocampal cholinergic system is relatively normal. For example, patients with Huntington’s disease (89) and Parkinson’s disease (90, 91) have intact eyeblink conditioning.
We combined the data over several studies from 61 probable AD patients and 100 normal age-matched older adults that we had tested in the 400-ms delay eyeblink conditioning procedure. To these data we applied a bivariate logistic regression analysis using percentage of CRs to predict diagnosis (92). With percentage of CRs as the only predictor, the area under the receiver operating characteristic curve (AROC) was 73%. Prediction was improved when age and gender were added to the model. In a multivariable logistic regression model including age, gender, and percentage of CRs, the model accurately distinguished patients with and without probable AD with AROC of 79.8%. Both the bivariate and the multivariable logistic models were statistically significant (p < 0.0001). The 400-ms delay eyeblink classical conditioning paradigm has utility in identifying patients with AD.
We have used the delay eyeblink conditioning paradigm on the basis that the hippocampus modulates acquisition of CRs in the cerebellum. When the hippocampal cholinergic system is disrupted, the cerebellum is disrupted and acquisition is delayed. But some have argued that since the trace eyeblink conditioning paradigm is hippocampus dependent, it should be a more sensitive test of AD. Before Clark and Squire (93) demonstrated that the trace had to be 1,000 ms long for the paradigm to be hippocampus dependent in humans, we tested patients with AD in the 750-ms trace paradigm (Fig. 1.1) and compared performance to the 400-ms delay paradigm (94). Though AD patients acquired a lower percentage of CRs in the 750-ms trace paradigm relative to controls, AD patients performed better in the 750-ms trace paradigm than in the 400-ms delay paradigm. We are not aware that AD patients have been tested in a long-trace paradigm, which is difficult to implement because the human spontaneous eyeblink rate is around one blink per second. It becomes difficult to determine which responses are CRs and which are spontaneous eyeblinks.
Longitudinal results suggest that 400-ms delay eyeblink conditioning has utility in the early detection of dementia. A 3-year longitudinal study of non-demented adults tested on eyeblink conditioning revealed that three normal subjects testing in the AD range (producing less than 25% CRs) at Time 1 became demented within 2 or 3 years (95). A fourth “normal” subject who scored just above criterion (26% CRs) also developed dementia within 2 years of the initial testing. A fifth subject in this group became demented and died within a year of the initial testing. Thus, of eight non-demented subjects age-matched to probable AD patients who scored on eyeblink conditioning in the AD range, only three were cognitively normal at the end of a 3-year period and 63% became demented within 2–3 years. Age-matched non-demented subjects scoring above criterion remained cognitively intact during the period of the longitudinal investigation. A second longitudinal study followed 20 cognitively normal elderly participants (half good-conditioners; half poor-conditioners) over a 2-year period (96). A neuropsychological test battery administered 2 years after the initial eyeblink conditioning test revealed significantly worse performance in poor conditioners on visuospatial abilities, semantic memory, and language abilities showing early decline in AD. Over a 2-year period, two of the poor conditioners (and none of the good conditioners) failed significantly, and one was diagnosed with probable AD.
1.2.1 Rabbits as Models for Alzheimer’s Disease
Rabbits are in the order Lagomorpha and have as separate ancestry from rodents. To assess the evolutionary relationships of Lagomorpha to other taxa, Graur and colleagues (97) used orthologous protein sequences. A total of 91 protein sequences were analyzed, and reconstructions revealed that primates are phylogenetically closer to lagomorphs than are Rodentia. From a phylogenetic standpoint, rabbits are closer than rodents to human primate ancestry.
From the standpoint of the Aβ proteins that are the central pathologic processes associated with neurodegeneration in AD, rabbit models have a definite advantage. The rabbit’s closer phylogenetic relation to primates is expressed in the amino acid sequence of Aβ, which is 97% identical to the human sequence (98). They form Aβ plaques. Species such as mouse and rat have a different amino acid sequence for Aβ and do not form these plaques.
As a species for cognitive assessment, rabbits have another advantage. On no other species is there such a large body of parametric data on the classically conditioned eyeblink response as on the rabbit (99). Much of the general literature on classical conditioning is based on data collected in the rabbit. From the early work of Hilgard in the 1930 s with human eyeblink conditioning, Gormezano in the 1960 s with rabbit eyeblink conditioning, and subsequent work of their colleagues and students, the parallels between rabbit and human eyeblink classical conditioning have been demonstrated empirically for decades.
It was the initial knowledge that eyeblink conditioning was impaired in normal aging in rabbits and humans that made this well-characterized model attractive for the investigation of neurobiological substrates of age-related memory impairment. The model system of eyeblink classical conditioning might be “the Rosetta stone for brain substrates of age-related deficits in learning and memory” (100). Both rabbits and humans show age-associated deficits in conditioning, and these can be easily dissociated from age-associated changes in sensory systems (i.e., differences in sensory thresholds for detection of the conditioned stimulus) or motor systems (differences in reflexive eye blink amplitude). Rabbits and humans also show parallel age-associated changes in the neural substrates critical for eyeblink classical conditioning, the cerebellum, and the hippocampus. Age-associated deficits can be artificially induced in both young rabbit and young human subjects with drugs that affect cholinergic neurotransmission, and age-associated deficits can be reversed in rabbits with drugs approved to treat cognitive impairment in AD (101). The fact that the same classical conditioning paradigm can be tested in both rabbits and humans makes eyeblink conditioning a valuable preclinical test of cognition-enhancing drugs.
One of the strengths of using eyeblink conditioning as a model of age-associated deficits in learning and memory is that both humans and rabbits show similar deficits as they age. Humans begin to show age-associated deficits in eyeblink conditioning at about 40–50 years of age (79, 102). Rabbits begin to show age-associated deficits at approximately 2 years of age (103). Based on declines in reproductive capacity, a 2-year-old rabbit is equivalent to a 40–45-year-old human, which suggests similar onset of age-associated declines in eyeblink conditioning in both humans and rabbits. Furthermore, the results found with rabbits appear to generalize to other non-human species, such as mice, rats, and cats.
The combined advantages of rabbits as a research species are numerous. Rabbits have a closer phylogenetic proximity to primates than do rodents, rabbits have an extensively characterized profile on a measure of learning and memory that closely parallels human performance, and rabbits and humans have a documented profile of age-related parallels in learning impairment. The model system of rabbit eyeblink conditioning has been used extensively to elucidate mechanisms of learning and memory in normal aging. Age is currently the best predictor of AD in humans.
1.2.1.1 The Cholesterol-Fed Rabbit as a Model of Alzheimer’s Disease
Vascular risk factors such as hypertension, type 2 diabetes, abnormal lipid levels, and obesity are risk factors common to Alzheimer’s and cardiovascular disease. Observations of Aβ-containing senile plaques in the brains of human patients with cardiovascular disease led Sparks and colleagues (104) to examine the heretofore unexplored brains of a long-standing animal model of heart disease, the cholesterol-fed rabbit.
An extensive elaboration of this rabbit model of AD has demonstrated that the cholesterol-fed rabbit is a valid model of AD on a number of levels. At a molecular level, the brain of this animal model has numerous (>12) features similar to the pathology observed in the AD brain including elevated Aβ concentration, neuronal accumulation of Aβ immunoreactivity, extracellular Aβ plaques, reduced levels of acetylcholine, elevated brain cholesterol, apolipoprotein E immunoreactivity, breaches in the blood–brain barrier, microgliosis, and neuronal loss.
Sparks also discovered that increased levels of Aβ in the brains of cholesterol-fed rabbits were dependent on the quality of the water the animals were administered. Animals given tap water accumulated considerably more Aβ in the brain than animals administered distilled water (105). It was in a collaborative attempt to demonstrate cognitive validity of cholesterol-fed rabbits that Sparks discovered that it was trace amounts of copper in the drinking water that were responsible for the higher levels of Aβ in the brain in comparison to the brains of rabbits administered the cholesterol diet and distilled water. In this issue, he elaborates his position that cholesterol in the diet causes accumulation of Aβ, whereas copper introduced in the drinking water impairs elimination of Aβ.
1.2.1.2 Cholesterol-Fed Rabbits, Eyeblink Conditioning, and Therapeutic Interventions
Sparks and Schreurs’ (106) demonstration that eyeblink classical conditioning was impaired in cholesterol-fed rabbits, in the light of our own work on eyeblink conditioning in human AD and classical conditioning in normal aging rabbits and humans, was a stimulating discovery that led us to initiate work with AD model rabbits. Our aim was to replicate AD neuropathology in the cholesterol-fed rabbit model, test eyeblink conditioning in this model, and determine if galantamine (Razadyne®) would ameliorate impaired conditioning. Rabbit chow with 2% cholesterol and drinking water with 0.12 ppm copper sulfate were administered for 10 weeks. Control rabbits received normal food and distilled water. Rabbit brains were probed for neuropathology. AD model rabbits had significant neuronal loss in frontal cortex, hippocampus, and cerebellum. Changes in neurons in the hippocampus were consistent with neurofibrillary degeneration and cytoplasmic immunoreactivity for β-amyloid and tau. In a second experiment, cholesterol-fed model rabbits were injected daily with vehicle or 3.0 mg/kg galantamine and tested on 750-ms trace and delay eyeblink conditioning (animals were conditioned first in trace, then in a delay paradigm matched for the ISI between CS and US onset). Galantamine improved eyeblink conditioning significantly over vehicle. We concluded that this animal model of AD has validity from neuropathological to cognitive levels and offers a promising addition to the available animal models of AD. Galantamine ameliorated impaired eyeblink conditioning, extending the validity of the model to treatment modalities (107).
One of the results highlighted in our report was the evident loss of Purkinje neurons and staining for Aβ and tau in the cerebellum of cholesterol-fed rabbits (confirmed by Sparks, unpublished observations). We examined the cerebellum in cholesterol-fed rabbits primarily because the cerebellum is the essential substrate for eyeblink classical conditioning, a behavior on which they were impaired. Although the cerebellum has traditionally been viewed as one of the last structures to be impacted by classical AD pathology in humans, evidence has mounted to indicate that the cerebellum is indeed affected by AD (108).
Sparks and Schreurs (106) had tested trace eyeblink conditioning in cholesterol-fed rabbits because medial temporal lobe structures affected in AD are essential for trace conditioning. In their study, rabbits tested in a short-delay paradigm were not impaired. We used the same interval between the tone conditioned stimulus and airpuff unconditioned stimulus in the delay and trace paradigms and found both to be impaired (see Fig. 1.2 for a comparison of a representative control rabbit (a) and a representative cholesterol-fed rabbit (b) trained in the 750-ms trace eyeblink conditioning paradigm). However, both Sparks and Schreurs (106) and our study tested the trace paradigm first. We followed up with a study of cholesterol-fed rabbits to determine if delay eyeblink conditioning would be impaired when tested before trace conditioning (109). Cholesterol-fed rabbits were significantly impaired in the 750-ms delay eyeblink conditioning paradigm (Fig. 1.1) when that paradigm was tested first, after 10 weeks of the 2% cholesterol and trace copper diet. Just as humans patients diagnosed with AD are significantly impaired in delay eyeblink conditioning, so are cholesterol-fed rabbits.
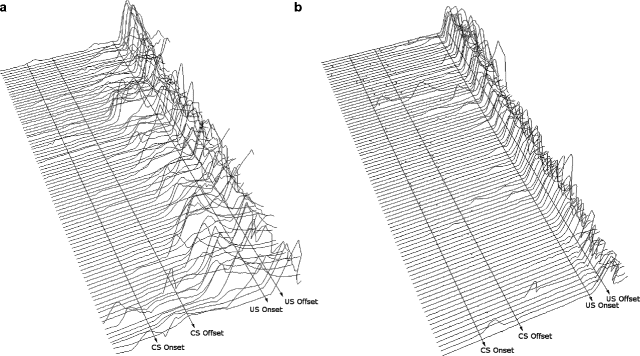
Fig. 1.2.
(a) and (b): Eyelid/nictitating membrane movement in rabbits as recorded from changes in the reflectance from the cornea (of the left eye) detected by infrared. Each line represents activity from an individual trial (1–90, with Trial 1 represented at the bottom of each figure and Trial 90 represented at the top of each figure) from Session 7 of 750-ms trace eyeblink classical conditioning. Total trial length is 1,300 ms. Lines are drawn to approximate the onset and offset of both the tone conditioned stimulus (CS) and the airpuff unconditioned stimulus (US). There are 250 ms in the pre-CS period before CS onset, the CS duration is 250 ms, the stimulus-free “trace” interval following CS offset is 500 ms, US duration is 100 ms, and there are 200 ms in the post-US period. A response is scored if eyelid or nictitating membrane movement meets or exceeds 10% of the maximum response generated in that session. Performance is shown for a young male rabbit on a standard diet (control rabbit; a) and a young male rabbit following 10-week exposure to a high (2%) cholesterol diet with distilled water supplemented with trace amounts of copper (Alzheimer’s model experimental rabbit; b). Each subject is representative of their respective groups. The incidence of conditioned responses (CRs) is significantly greater in the control subject (a) relative to the Alzheimer’s model rabbit (b).
2 Materials
2.1 Human Eyeblink Equipment
2.1.1 Apparatus
The eyeblink conditioning apparatus is commercially available from San Diego Instruments and consisted of a portable, automated system with an infrared eyeblink detector and airpuff jet attached to headgear and input to an interface box containing a microprocessor and a miniature air compressor. The interface box is interactive with a computer. The headgear has an adjustable headband holding the airpuff jet and infrared eyeblink monitor. Filtered air is compressed in the interface box and delivered through a jet set approximately 2 cm from the cornea.
The voltage from the infrared device is amplified and differentiated; eyelid data are transferred to the computer for storage and analysis. From the speaker of the interface box, a 1,000-Hz, 80-dB sound pressure level (SPL) tone CS is delivered through headphones. Air for the corneal airpuff US is compressed in the interface box, filtered, and delivered at a pressure of 5–10 psi. The timing of stimulus presentations is controlled by the microprocessor in the interface box.
2.2 Rabbit Eyeblink Equipment
2.2.1 Eyeblink Classical Conditioning
The conditioning apparatus consists of four separate sound-attenuating chambers, permitting four rabbits to be trained simultaneously. A speaker mounted to the wall of each chamber delivers a pure tone (1 KHz, 85 dB SPL) used as the conditioned stimulus (CS). Each box has a fan running throughout the experiment that provides low-decibel background noise. The headpiece, affixed behind the rabbit’s ears and under its muzzle, holds a plastic tube to deliver 7–8 psi corneal-directed airpuff unconditioned stimulus (US) and an infrared emitter and detector (San Diego Instruments, San Diego, CA) to measure the rabbit’s eyeblink response. This device is positioned approximately 2 cm from the rabbit’s eye. Eyelid retractors keep the rabbit’s eye open. Output from the infrared detector is stored and analyzed using a PC. This system also controls the timing and presentation of the stimuli. The intertrial interval is randomized and ranges between 20 and 30 s. A single session lasts approximately 45 min and consists of 90-paired CS–US trials.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


