Chapter 7 Evaluation of Hemostasis
Coagulation and Platelet Disorders
Blood Platelets (Thrombocytes)
Normal Morphology
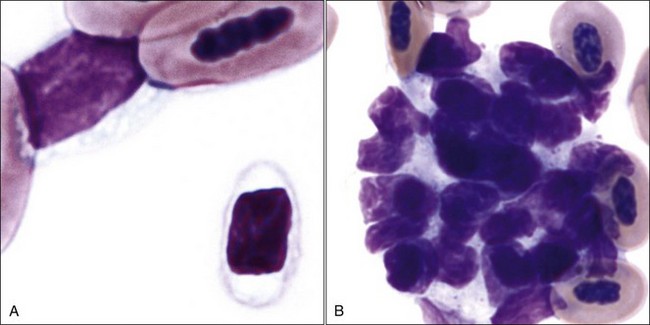
Blood platelets (thrombocytes) in mammals are small round-to-oval anucleated cell fragments (thin discs when unstimulated) that form from proplatelet cylinders of megakaryocyte cytoplasm (see Chapter 3). Platelet cytoplasm appears light blue, with many small reddish-purple granules when visualized using routine blood stains (Fig. 7-1, A,B). Equine platelets often stain poorly with Wright-Giemsa stain (Fig. 7-1, C), but they generally stain better with Diff-Quik (Fig. 7-1, D).
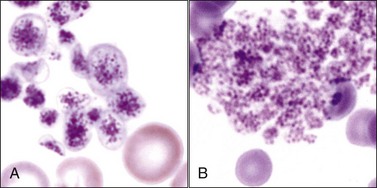
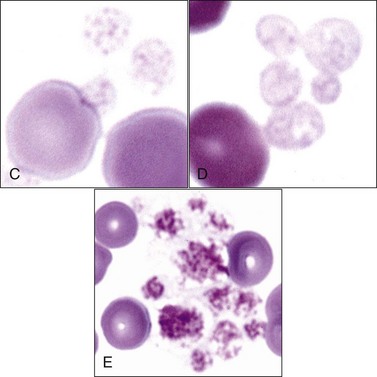
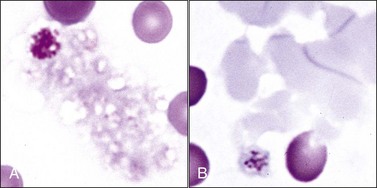
Domestic cats (and other members of the Felidae family) normally exhibit greater variation in platelet size (2 to 6 µm or larger) than seen in other domestic animals or humans (2 to 4 µm). This size variability in cats may be related to an altered M-loop region in β1-tubulin compared with other mammals.49 The presence of these larger platelets (Fig. 7-1, E) results in higher mean platelet volumes (MPVs) in healthy cats than in other domestic animals. Feline platelets appear especially sensitive to activation during blood sample collection and handling, resulting in degranulated platelet aggregates, which may be overlooked by an inexperienced observer (Fig. 7-2, A). Some of the precipitated cryoglobulin recognized in blood from a cat with a monoclonal cryoglobulinemia has also been found to resemble aggregates of degranulated platelets (Fig. 7-2, B).237 Platelets typically stain uniformly purple with the new methylene blue wet preparation (Fig. 7-3).
Newly formed platelets have higher RNA content and have been termed reticulated platelets. They cannot be quantified by morphology but can be counted using flow cytometry following labeling of RNA with a fluorescent dye.400,442,542
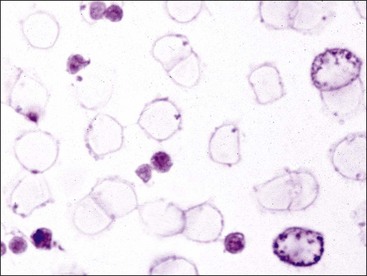
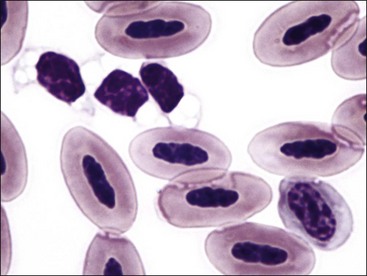
Thrombocytes in nonmammalian species have nuclei and are much larger than those of mammals. They have a high nuclear-to-cytoplasmic (N : C) ratio and are often oval or elongated with light blue or nearly colorless cytoplasm in stained blood films (Figs 7-4, A, 7-5). Cytoplasmic vacuoles may be present at one or both ends of a cell. Granules are usually not apparent. When thrombocytes are more round in shape, they can be difficult to differentiate from lymphocytes. Like mammalian platelets, thrombocytes appear clumped on blood films when they are activated (Fig. 7-4, B).
Platelet Life Span and Counts in Blood
Mean platelet life spans of 4 to 6 days have been reported in dogs, cats, horses, and cattle.121,231,257,470 The platelet life span increased from a mean of 5.5 days to a mean of 8 days after splenectomy in dogs, suggesting that the spleen is especially adept at recognizing and removing aged platelets in dogs.124,319 Platelet senescence is associated with changes that have characteristics of apoptosis, including increased surface phosphatidylserine (PS), which may promote their removal from circulation by macrophages.409
Normal platelet counts vary depending on the species, with minimal reference values as low as 100 × 103/µL in horses and maximal reference values as high as 800 × 103/µL in several domestic animal species.260 The numbers normally present in blood greatly exceed those needed for adequate hemostasis. Thrombocyte counts are much lower in nonmammalian species than they are in mammals. In most avian species, thrombocyte counts range between 20 × 103/µL and 30 × 103/µL.221
Platelet Metabolism
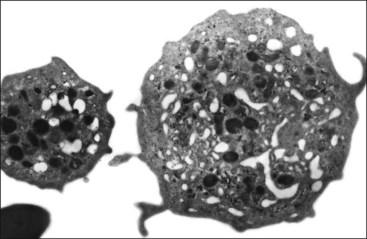
As in other blood cells, glucose is the major energy source for platelets. In contrast to anucleated erythrocytes, platelets have mitochondria and consequently utilize the Krebs cycle and oxidative phosphorylation. Little of the pyruvate produced by glycolysis is metabolized through the Krebs cycle in resting platelets, but oxidative metabolism, as well as glycolytic metabolism, is markedly increased when platelets are activated to meet increased needs for adenosine triphosphate (ATP) production. Biochemically, platelets have often been compared with white skeletal muscle. They have active anaerobic glycolysis and synthesize and utilize large amounts of glycogen.402 Platelets have a dense tubular system analogous to the calcium ion (Ca2+) sequestering sarcoplasmic reticulum present in skeletal muscle (Fig. 7-6). Except for ruminants and horses, domestic mammals have a well-developed canalicular system that is continuous with the surface membrane (Fig. 7-7).46 Microtubules and microfilaments are present and composed of a variety of contractile proteins including actin, myosin, and related proteins. Microtubular coils help to maintain the discoid shape of resting platelets.46 Changes in actin filament conformation and the organization of associated proteins are required for platelet shape change, spreading, aggregation, secretion, and clot retraction.46
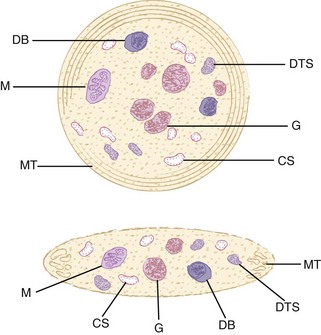
Adenine nucleotides are present in metabolic and storage pools within platelets. As in other cells, ATP accounts for most of the adenine nucleotide in the metabolic pool. The ATP in this pool provides energy for cell functions. In addition to energy needed for normal homeostatic processes, platelets expend large amounts of energy during the release reaction and aggregation, to be discussed later. The storage pool of adenine nucleotides is contained within dense granules (also called dense bodies or δ-granules), which contain about two-thirds of the total adenine nucleotides in the cell. In contrast to the cytoplasm, where ATP predominates, the ADP/ATP ratio is about 1.5 in dense granules. In addition to adenine nucleotides, these dense bodies contain considerable amounts of serotonin, Ca2+, inorganic polyphosphates, and smaller amounts of other molecules, which are all secreted outside the platelets during the release reaction (degranulation).427 The contents of α-granules and some of the contents of lysosomes are also secreted when platelets are activated. Some of the contents of α-granules are synthesized by megakaryocytes and others are taken up from the plasma. Although α-granule content varies by species, a partial list might include adhesive proteins (von Willebrand factor [vWF], fibrinogen, fibronectin, thrombospondin), coagulation factors (factors V, VII, XI and XIII), protease inhibitors (plasminogen activator inhibitor, tissue factor pathway inhibitor, α2-antiplasmin), CXC chemokines (platelet factor 4 [PF4] and β-thromboglobulin), P-selectin (CD62P), glycoprotein (GP) receptors GPIb/IX/V (CD42) and GPIIb/IIIa (CD41/CD61), and other components, including chemotactic, mitogenic, and vascular permeability factors. Platelets also contain lysozyme granules, which contain hydrolytic enzymes.46,57,427
Platelets express various glycoprotein molecules on their surfaces that are needed for normal adhesion (platelet to extracellular matrix) and aggregation (platelet-to-platelet binding). The glycoprotein complex GPIb/IX/V is especially important for the adhesion of platelets to vWF that is bound to the subendothelial matrix. The GPIIb/IIIa complex (αIIbβ3 integrin) is essential for normal platelet aggregation, which is mediated largely by fibrinogen.271
Platelets have several functions in hemostasis. The first is the formation of a platelet plug at the site of vessel injury. Formation of a platelet plug alone is sufficient to stop bleeding from an injury to a small vessel. Second, platelet activation results in the translocation of negatively charged phospholipids (primarily PS) from the inner surfaces to the outer surfaces of platelets. These aminophospholipids bind certain coagulation factors in close proximity on platelet surfaces, thereby accelerating coagulation.271 Third, activated platelets secrete PF4 (CXCL4) from their α-granules, which binds to heparin-like molecules on endothelial surfaces near the activated platelets. This binding displaces antithrombin (also called antithrombin III), which also binds to these heparin-like molecules, thereby inhibiting local antithrombin activity and promoting coagulation.7
Finally, the presence of platelets helps maintain normal vascular integrity in some manner. Vascular endothelium is thin, fragile, and leaky in animals with low platelet counts (thrombocytopenia).284 Platelets help protect vessels from bleeding during inflammation in a way that is not dependent on the formation of a platelet plug.194 The relationship between platelets and endothelial cells is complex and poorly understood, but platelet granules have a variety of vasoactive molecules that might promote the integrity of the endothelial barrier. Sphingosine-1 phosphate, a phospholipid product of platelets, is one substance that appears to be an important mediator protecting the vascular barrier.451
Primary Hemostasis
Vascular Phase
Primary hemostasis consists of a vascular phase and a platelet phase. Following the severing of vessels, a reflex vasoconstriction temporarily retards blood flow, allowing time for formation of the platelet plug to begin and coagulation to commence. The damage or removal of endothelial cells exposes the subcellular matrix, resulting in platelet adhesion. Following vessel injury, tissue factor (TF) on subendothelial adventitial cells activates coagulation.468 Although platelet plug formation and coagulation are discussed below in separate sections, it is important to realize that these processes are interrelated. Thrombin generated during coagulation is one of the agonists that activates platelets, and coagulation occurs on the surface of activated platelets in vivo, which greatly accelerates the coagulation process.271
Platelet Phase
In response to vessel wall injury or exposure to foreign surfaces, platelets rapidly undergo the processes of adhesion, shape change, secretion, and aggregation through a complex series of coordinated processes that culminate in the formation of a precisely located platelet plug. Because of the rapidity of intracellular responses and synergy between secondary messenger systems, it is not possible to organize the various effector systems into a clearly defined temporal sequence. Reciprocal activation of effector systems also makes it difficult to define a clear sequence of events.271
Platelet Adhesion
Optimal platelet adhesion requires the binding of the platelet surface glycoprotein GPIb/IX/V complex to vWF molecules on the surface of the subendothelium. The GPIb/IX/V complex does not bind circulating vWF; it binds only vWF molecules that have been immobilized within the extracellular matrix, where vWF is bound primarily to collagen. It appears that the conformation of vWF changes upon binding to the extracellular matrix, so that it readily binds to the GPIb subunit of the GPIb/IX/V complex on platelets, especially when shear force is applied, as occurs when flowing blood is exposed to the subendothelial surface (Fig. 7-8).271
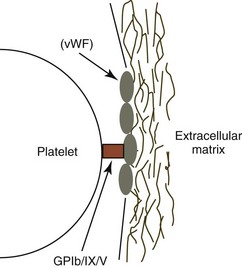
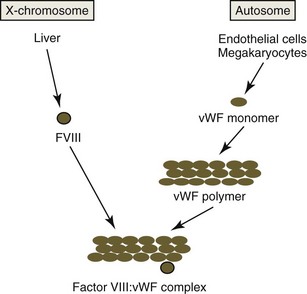
vWF is a component of the factor VIII : vWF macromolecular complex (Fig. 7-9). It circulates as a series of disulfide-bonded polymers with a molecular weight of up to 20 × 103 kDa in blood. vWF polymers bind to factor VIII (FVIII), a smaller protein (molecular weight 285 kDa) that functions as a procoagulant.79 In humans, about 1 molecule of FVIII binds per 50 monomers of vWF. Binding of FVIII to vWF prolongs the circulation time of FVIII.513 These two factors are controlled by different genes and are synthesized independently. vWF, previously referred to as factor VIII : R (related antigen), is encoded by an autosomal gene and synthesized by endothelial cells and megakaryocytes (in some species). FVIII, also referred to as antihemophilic factor, is an X-linked gene product that is synthesized primarily in the liver (probably by hepatocytes).309 The binding of vWF to the extracellular matrix is associated with decreased binding of FVIII.513
Although vWF appears to be the most important factor in the extravascular matrix for platelet adhesion under flow conditions, other factors can promote platelet adhesion. Thrombospondin-1 is an alternate substrate to vWF for binding to platelet GPIb under high shear conditions. Under static conditions with low shear, platelets adhere directly to collagen in the subendothelium using GPVI and α2β1 (GPIa-IIa) receptors.271
Platelet Activation
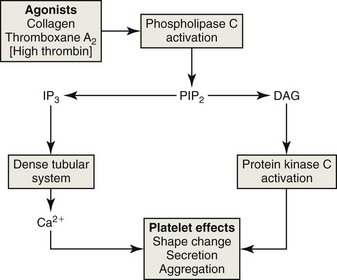
The adhesion of platelets to the extracellular matrix under shear conditions and their binding to collagen and other strong agonists, including thrombin generated during coagulation, result in the activation of phospholipase C (PLC) in the platelet membrane (Fig. 7-10). PLC cleaves a unique membrane phospholipid, phosphatidylinositol, 4,5-bisphosphate (PIP2), into inositol triphosphate (IP3) and 1,2-diacylglycerol (DAG). IP3 diffuses through the cytoplasm and binds to a calcium channel receptor in the dense tubular system, which stimulates the release of calcium ions.271 The rise in the cytosolic concentration of calcium results in a cascade of intracellular changes and activity. Calcium ions and DAG bind to and activates calcium diacylglycerol guanine nucleotide exchange factor I (CalDAG-GEFI), which activates the Rap1b GTPase through the exchange of bound GDP for GTP. Activated Rap1b promotes the conformational change and activation of the GPIIb/IIIa integrin, so that it binds fibrinogen. CalDAG-GEFI also appears to link increases in intracellular calcium ions to signaling pathways that regulate the synthesis of thromboxane A2 (TxA2) and release of granules.33 Inherited defects in CalDAG-GEFI lead to defects in platelet function and to bleeding in affected dogs and cattle.44 Intracellular calcium ions and DAG together work to activate protein kinase C isoforms, which phosphorylate other molecules. Taken together, these reactions and others, including phosphatidylinositol-3 kinase (PI3K), stimulate platelets in various ways, resulting in TxA2 synthesis, shape change, integrin activation, secretion, aggregation, and platelet procoagulant activity.66,191,220
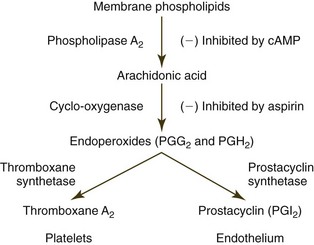
Phospholipase A2 is also activated, which stimulates the hydrolysis of phospholipids (especially phosphatidylcholine) from the dense tubular system, causing the release of arachidonic acid. Arachidonic acid is subsequently metabolized to TxA2 by the cyclooxygenase enzyme pathway (Fig. 7-11).271 Phospholipase A2 (in conjunction with an acetyltransferase) is also involved in generating platelet-activating factor (PAF, 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine), another agonist that induces platelet aggregation.66,102 PAF can be produced not only by activated platelets but also by activated endothelial cells and leukocytes.113,298,533 PAF is an important proinflammatory mediator that has multiple effects beyond platelet activation.218,298,314
Epinephrine does not function as an agonist by itself but potentiates platelet activation and secretion induced by other agonists. Platelets from some dogs do not respond to TxA2 in vitro owing to impaired TxA2-G protein coupling, but this defect is normalized by pretreatment with epinephrine.44
Platelet activation is inhibited by prostacyclin (prostaglandin I2, or PGI2). When stimulated by agonists including thrombin and PAF, endothelial cells produce increased amounts of PGI2 as a product of arachidonic acid metabolism (see Fig. 7-11).234,406 This antagonist has an antiaggregatory effect by stimulating cyclic adenosine monophosphate (cAMP) synthesis in platelets. Agonists tend to counteract this effect by lowering platelet cAMP concentrations.264,271 Activated endothelial cells also suppress platelet reactivity by increasing the rate of nitric oxide (NO) synthesis. NO inhibits platelet activation by activating intracellular guanylate cyclase, which leads to the formation of cyclic guanosine monophosphate (cGMP).271 NO and PGI2 also function as potent vasodilators.264,356
Change in Platelet Shape
The discoid shape of resting platelets is maintained by circumferential bundles of microtubules beneath the platelet membrane and an extensive network of short actin filaments, forming a membrane skeleton. Following binding to vWF and collagen, platelets are activated, the microtubular coils reorganize into linear arrays, and actin disassembles and reassembles as platelets form filopodia and spread.46,271 Negatively charged phospholipids and glycoprotein receptors are also exposed on the surface. Platelets have surface receptors for various subendothelial components including collagen, fibronectin, laminin, and thrombospondin.31 The collagen receptor (α2β1 integrin) functions both in adhesion and as an agonist receptor that activates platelets.46
Platelet Secretion
Platelet secretion (release or degranulation) requires energy-dependent contractile mechanisms. Granules are crushed together (fusion and dissolution) by a surrounding web of microtubules and microfilaments. The contents of dense bodies and granules are discharged into the open canalicular system, which is continuous with the platelet surface.427 Ruminant, equine, and elephant platelets have minimal canalicular systems; granules and dense bodies primarily discharge their contents by fusing with the external platelet membrane.46,145 Contraction of individual platelets and the platelet aggregate facilitates the discharge of material into surrounding plasma. ADP, serotonin, and calcium released from dense bodies promote platelet aggregation.427
Platelet Aggregation
ADP, TxA2, thrombin, and PAF are important agonists that promote platelet aggregation. Serotonin is also an important agonist in some species. Optimal platelet aggregation requires fibrinogen and Ca2+. The actions of these agonists result in the increased exposure and activation of GPIIb/IIIa, a β3-integrin platelet surface receptor that binds to fibrinogen (Fig. 7-12). Aggregation occurs when symmetric fibrinogen molecules bind to exposed receptors on adjacent platelets. vWF also promotes platelet aggregation when shear forces are high in flowing blood.271,462 The resultant platelet plug may be sufficient to stop bleeding from small vessels. As discussed further on, the bleeding-time test, which measures the time needed to form the platelet plug, is dependent on platelet numbers and function.
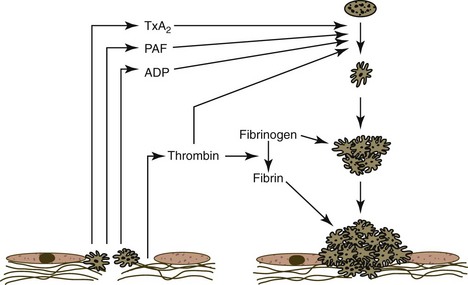
Platelet Procoagulant Activity
Phospholipids are asymmetrically located in the plasma membrane of resting cells, including platelets, because of the presence of the Mg-ATP-dependent enzymes flippase and floppase. Negatively charged aminophospholipids are concentrated more in the inner leaflet of the platelet membrane because of an inward-directed pump for PS and phosphatidylethanolamine known as aminophospholipid translocase or flippase. Neutral phospholipids, including phosphatidylcholine and sphingomyelin, are transported to the outer membrane leaflet of platelets by floppase activity.77
The activation of platelets by agonists results in the calcium-dependent activation of scramblase (an enzyme that promotes unspecific bidirectional redistribution of phospholipids across the bilayer) and inhibition of flippase activities. The net result is that negatively charged phospholipid (primarily PS) is translocated from the internal leaflet to the external leaflet of the plasma membrane.173,279 Negatively charged aminophospholipids on the surface of activated platelets have been termed “platelet factor 3” or “platelet procoagulant activity.” The translocation of PS to the surface of activated platelets accelerates coagulation because positively charged Ca2+ binds to the negatively charged phospholipids and to the negatively charged carboxyl groups of coagulation factors. Binding of coagulation factors to platelets not only brings them together to enhance interactions but also helps to protect them from inhibitors.271
Platelet activation results not only in enhanced reactivity toward other platelets—forming platelet aggregates (Fig. 7-13) and prompting enhanced reactivity toward leukocytes, thus forming platelet-leukocyte aggregates—but also in the shedding of microparticles (microvesicles) from the platelet surface.365,545 These microparticles have procoagulant activity, expressing PS, activation-dependent adhesion molecules, and possibly TF on their surfaces.279,381 Platelet microparticles bind to and activate leukocytes and endothelial cells, which may promote thrombosis and inflammation.77,187 Platelet activation ultimately results in platelet changes that appear to be proinflammatory and broadly consistent with cell necrosis.255
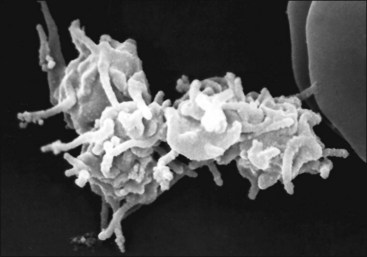
PS is also exposed on the surface of platelets undergoing apoptosis via a pathway different from that for platelet activation. This PS exposure presumably promotes platelet clearance under steady-state conditions. Whether this has any relevance to thrombin generation in vivo remains to be determined.255
Secondary Hemostasis
Overview
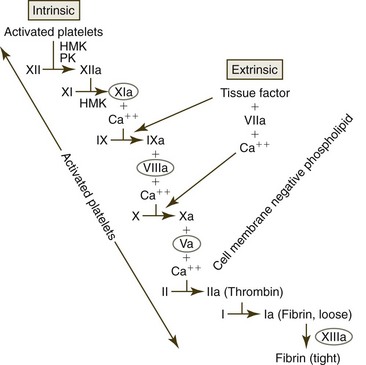
Coagulation has classically been divided into intrinsic and extrinsic coagulation pathways (Fig 7-14), with both utilizing a common pathway beginning with factor X (FX). This presentation of coagulation events is useful in dividing coagulation into components in order to study their relationships and interpret diagnostic test results, but it is not accurate from a physiologic point of view. The classic presentation implies that coagulation can be induced by either the extrinsic or the intrinsic pathway. It does not show the importance of the extrinsic pathway in activating the intrinsic pathway or that coagulation is a cellular or at least membrane-bound event.368
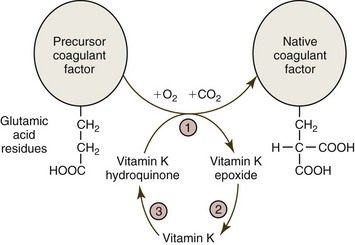
Vitamin K is required for the synthesis of functional factors II, VII, IX, and X, as well as protein C and protein S. Following synthesis of the protein molecules in the liver, a vitamin K-dependent carboxylation (via γ-glutamyl carboxylase) of glutamic acid residues on these molecules is required for them to bind Ca2+ and become functional (Fig. 7-15). Cats and sheep that have γ-glutamyl carboxylase deficiencies have bleeding episodes because of low vitamin K-dependent coagulant factor activities.267,473 Dicumarol-type anticoagulant drugs, including warfarin and brodifacoum, inhibit the vitamin K epoxide (or oxido) reductase (VKOR) enzyme complex needed to recycle vitamin K, resulting in the production of inactive coagulation proteins.105,477
Initiation of Coagulation (Activation of the Extrinsic Pathway or Tissue Factor Pathway)
A small amount (about 1%) of factor VII (FVII) in blood is activated (FVIIa). Coagulation is initiated in vivo when blood containing FVIIa comes in contact with TF on the surface of cells or cell membranes (i.e., microparticles from cells) (Fig. 7-16).77 TF is a glycoprotein that depends on its association with negatively charged phospholipid (most notably PS) to bind FVII/FVIIa. TF is not expressed on unstimulated blood or endothelial cells but it is constitutively expressed on the surface of many other cell types that are not normally in contact with the circulation. This includes cell types surrounding vessels as well as parenchymal cells in a variety of tissues, such as brain, heart, lung, and kidney.405
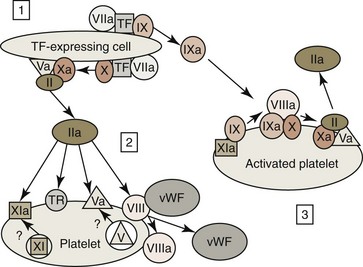
Following vessel injury, TF on subendothelial adventitial cells—such as adventitial fibroblasts, pericytes, and smooth muscle cells—activates coagulation. TF expression can also be induced in blood monocytes and possibly endothelial cells by endotoxin and/or inflammatory cytokines. TF has been reported on activated platelets; but, if present, this may come from the transfer of microparticles from activated monocytes or other cells to platelets.173,397
The TF-FVIIa complex on membrane surfaces activates additional amounts of FVII to FVIIa, which generates larger amounts of the TF-FVIIa complex on membranes. This complex activates factor IX (FIX) and FX to FIXa and FXa respectively. Although not optimal, FXa can directly activate some factor V (FV) to FVa, and the combination of FXa with cofactor FVa forms a complex called prothrombinase on the surfaces of TF-bearing cells, which converts prothrombin to thrombin. The activation of this tissue factor pathway in vivo functions to rapidly generate trace amounts of thrombin; however, the small amount of thrombin generated results in minimal fibrin formation. Coagulation generally begins on the surface of TF-bearing perivascular cells, but it is completed on the surface of activated platelets.467
Amplification of Coagulation
Following vessel injury, platelets bind via vWF to the subendothelial extracellular matrix near the TF-bearing cells that activate coagulation. Platelets are activated by this binding under shear conditions. Thrombin produced by activation of the tissue factor pathway also activates platelets by binding to thrombin receptors (see Fig. 7-16), including protease-activated receptors (PARs) and GPIb/IX/V.44 Following activation, platelets produce agonists—including TxA2, PAF, and ADP—which, together with thrombin, induce platelet aggregation and platelet plug formation. Platelet activation also results in the exposure of negative phospholipids (platelet procoagulant) on the platelets’ surfaces. Some coagulation factors in platelet granules (including FV and FXI, at least in some species) are also deposited on platelet surfaces after platelet activation and secretion.
The vWF/FVIII complex binds to activated platelets, and thrombin cleaves a portion of the FVIII molecule to form activated FVIIIa. FVIII activation results in the dissociation of this molecule from vWF.375 FVIIIa remains bound to platelets because activated platelets express distinct binding sites for FVIIIa (see Fig. 7-16). Thrombin also activates FV to FVa. This cofactor not only circulates in blood but may also be present in platelet granules and may be deposited on activated platelet surfaces following granule secretion.244 Finally, thrombin activates FXI to FXIa, which is also bound to the surfaces of activated platelets.79,137 At the end of the amplification phase of coagulation, activated platelets have negatively charged phospholipids, FVa, FVIIIa, and FXIa, present on their surfaces, priming them for rapid propagation of coagulation (see Fig. 7-16).243
Propagation of Coagulation (Activity of the Intrinsic Pathway)
Factor XII Activation of Factor XI
A long-held consensus mechanism for the activation of coagulation in vitro (contact activation) is shown in Figure 7-17.182,514 In this model, factor XII (FXII) comes in contact with an anionic, wettable surface (including blood collection tubes made of glass) and undergoes autoactivation, which generates a small amount of active FXII (FXIIa). FXIIa may autohydrolyze FXII to produce more FXIIa. In addition, FXIIa interacts with prekallikrein, which is bound to high-molecular-weight kininogen (HMK). Reciprocal interactions between FXIIa and prekallikrein generate an active fragment of FXIIa (FXIIf) and kallikrein respectively. FXIIa activates factor XI (FXI), which is also bound to HMK, which propagates the intrinsic pathway of coagulation.182 Some aspects of this model of contact activation (especially the interactions of these proteins with foreign surfaces) have been challenged.514
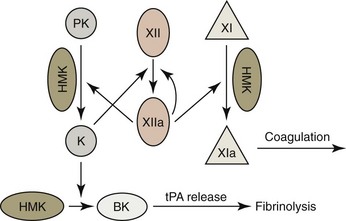
Because people, cats, and dogs with FXII deficiency exhibit no hemorrhagic tendencies and some marine mammals, birds, and reptiles naturally lack this factor, it has generally been accepted that FXII activation is of little importance to normal hemostasis in vivo.185 However, FXII is acknowledged to be important in the plasma kallikrein-kinin system. FXIIf-generated kallikrein converts HMK to bradykinin, which causes vasodilation, increased vascular permeability, and prompts an inflammatory response.265 Bradykinin also stimulates the release of tissue plasminogen activator (tPA) from endothelial cells, which promotes fibrinolysis, as discussed further on.182 FXIIf activates complement (C1r) in addition to generating kallikrein.275
Although FXII is apparently not essential for normal hemostasis, it may play a role in the growth of thrombi under pathologic conditions.100,428 The activation of human platelets results in the secretion of dense granule contents, which include polyphosphates. FXII appears to be activated following binding to polyphosphates on the surfaces of platelets. This FXII activation promotes both coagulation and the generation of bradykinin.265 Secreted polyphosphates may have additional effects on coagulation. Polyphosphates are reported to enhance thrombin-mediated FV activation, and their incorporation into fibrin fibers is reported to make them more resistant to lysis.80
Thrombin Activation of FXI
FXI binds to activated platelets via HMK and is believed to primarily be activated in vivo by thrombin initially generated by the activation of the tissue factor pathway. However, some FXI activation may also occur as a result of FXIIa on activated platelets, and FXI may be autoactivated by FXIa.80,154 Because FXI deficiency results in only mild bleeding in animals and humans (compared with the severe bleeding that occurs with FIX and FVIII deficiencies185) FXI does not appear to have a major role in early fibrin generation. FXIa is postulated to be part of a positive feedback loop that sustains thrombin generation through FIX activation to subsequently generate large amounts of fibrin.154 In addition, some FIXa generated on TF-bearing cells can detach and bind to nearby activated platelets and promote coagulation on platelet surfaces (see Fig. 7-16).271 Thus FIX is activated by both FXIa and the TF-FVIIa complex. In contrast, little of the FXa generated by TF-bearing cells becomes bound to activated platelets, because FXa that dissociates from the TF-binding cells is rapidly inactivated by the antithrombin and tissue factor pathway inhibitor (TFPI) present in blood. FIXa is not inactivated by TFPI and only slowly inactivated by antithrombin.467
FIXa combines with cofactor FVIIIa on activated platelet surfaces to form a complex sometimes called tenase that activates FX (Figs. 7-16, 7-18).185 FVa that is bound to platelet surfaces functions as a cofactor for FXa and forms a complex called prothrombinase on the surfaces of activated platelets. This complex converts large amounts of prothrombin to thrombin. The thrombin thus formed is released from platelets and converts fibrinogen to fibrin monomers; these polymerize spontaneously by hydrogen bonding to form unstable non-cross-linked fibrin polymers around the platelet plug (see Fig. 7-18).
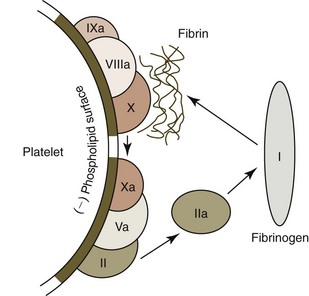
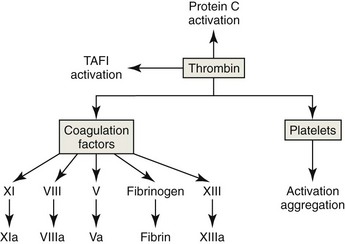
Stabilization of the Thrombus
The last step in fibrin polymerization involves the formation of covalent cross-links between fibrin monomers. Factor XIII (FXIII, fibrin stabilizing factor) is activated by thrombin. The FXIIIa formed is a Ca2+-dependent transglutaminase that catalyzes the formation of covalent bonds between lysine and glutamine residues of different monomers. Cross-linked fibrin is an insoluble protein polymer that stabilizes the platelet plug.185 Thrombin also activates a thrombin-activatable fibrinolysis inhibitor (TAFI) that makes the fibrin formed more resistant to lysis (see “Fibrinolysis,” below).328 Finally, the incorporation of polyphosphates from the dense granules secreted by platelets into fibrin fibers is reported also to make fibrin more resistant to lysis.80
Inhibitors of Thrombus Formation
Inhibition of Platelet Aggregation
Endothelial cells synthesize and release PGI2 and NO, powerful vasodilators that inhibit platelet aggregation. Endothelial cells also inhibit platelet function by virtue of an ectoenzyme (Ecto-ADPase/CD39/NTPDase) that has adenosine diphosphatase (ADPase) activity and can degrade ADP released from activated platelets.264
Antithrombin
Endothelial cells synthesize heparan sulfate proteoglycans, which are tightly associated with the endothelium and accelerate the inactivation of coagulation factors by antithrombin, the major thrombin inhibitor.461 In addition to thrombin, antithrombin inhibits other proteases, including factors IXa, Xa, XIa, and XIIa.185
Protein C Anticoagulant Pathway
Endothelial cells express the membrane protein thrombomodulin on their surfaces (Fig. 7-19). Thrombin binds to thrombomodulin, which inhibits thrombin’s procoagulant activity. This 1 : 1 thrombin : thrombomodulin complex activates both protein C and TAFI, discussed under “Fibrinolysis,” below. Protein C is a vitamin K-dependent plasma protein that circulates in an inactive form. It binds to an endothelial protein C receptor (EPCR), which enhances its activation by the thrombin : thrombomodulin complex. Activated protein C dissociates from EPCR and interacts with protein S (a vitamin K-dependent cofactor) to inhibit coagulation by proteolytically degrading FVa and FVIIIa on the surfaces of activated platelets. Although the physiologic significance of its effect is unclear, activated protein C can also promote fibrinolysis by inactivating the plasminogen activator inhibitor 1 (PAI-1).156,157
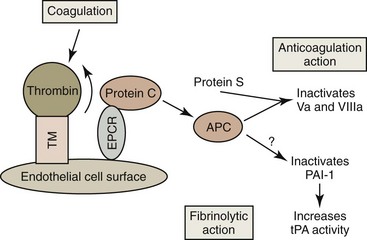
Tissue Factor Pathway Inhibitor
TFPI is synthesized by endothelial cells, and most TFPI is associated with the endothelium. FXa and the TF-VIIa complex are inhibited by the bivalent TFPI formerly called the lipoprotein-associated coagulation inhibitor.137 Protein S is a cofactor for the TFPI system, in addition to the protein C anticoagulant pathway.92
Platelet-Secreted Inhibitors
In addition to their potent roles in promoting coagulation, platelets secrete coagulation inhibitors, including protease nexin-1 (inhibits thrombin), protease nexin-2 (inhibits FXIa), and TFPI from α-granules. These factors may help to prevent the propagation of thrombosis beyond a site of injury.516
Fibrinolysis
Fibrinolysis is activated by the release of tPA from damaged endothelium. The amount of tPA available to stimulate fibrinolysis may be increased by activated protein C inactivating PAI-1.156 Plasminogen coprecipitates with fibrin as a thrombus forms. Conversion of plasminogen to plasmin by tPA is accelerated in the presence of fibrin (Fig. 7-20).377 Plasmin is not a highly specific enzyme, but its affinity for fibrin helps to limit its action. Plasmin-catalyzed hydrolysis of fibrin results in the formation of fibrin degradation products (fibrin split products), which have antihemostatic properties.185
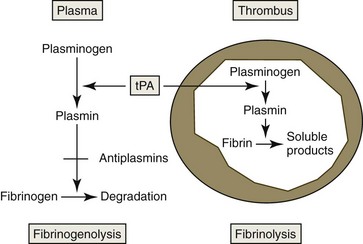
Inhibitors of fibrinolysis also occur in plasma. Endothelial cells produce PAI-1, which inhibits tPA. TAFI is activated by the thrombin-thrombospondin complex. It removes carboxy-terminal lysine groups from fibrin, resulting in decreased binding of plasminogen and tPA to fibrin and consequently decreased generation of plasmin. This slows but does not eliminate fibrinolysis.328,553 Thrombin’s central role in coagulation and fibrinolysis is summarized in Figure 7-21.
Fibrinolysis occurs more readily in capillaries than in the systemic circulation. A much higher density of endothelial cells is found in capillaries. Consequently thrombomodulin-mediated protein C activation and thrombin clearance are greater in capillaries than in large vessels.444 In addition, tPA release is greater in capillaries, and there may be less antiplasmin available to inhibit fibrinolysis. Rapid fibrinolysis of thrombi in large vessels could be life-threatening, but fibrinolysis is likely important in maintaining the integrity of capillary beds.
Anticoagulants
Coagulation is prevented in blood samples by using either Ca2+ chelators (ethylenediaminetetraacetate [EDTA] and citrate) or heparin in blood collection tubes. EDTA is the preferred anticoagulant for complete blood count (CBC) determinations in most species. Minimal sample dilution occurs following mixing with EDTA, and blood films prepared by using this anticoagulant exhibit optimal staining with routine blood stains. Blood from nonmammals has often been collected for hematology studies using heparin (rather than EDTA) as an anticoagulant because the blood of some birds (ratites) and some reptiles (chelonians) has been reported to hemolyze when collected with EDTA. This is clearly not a universal problem in birds and reptiles.222 The disadvantage of heparin is that leukocytes do not stain as well and platelets (thrombocytes) usually clump more than in blood collected with EDTA.
Sodium citrate has generally been the preferred anticoagulant for collecting plasma for coagulation tests and for collecting platelets for platelet function tests.84,363 However, hirudin (a leech-derived direct thrombin inhibitor) performed better than citrate as an anticoagulant in a whole-blood impedance aggregometer.25 Samples collected in citrate solution are diluted by 10%. If platelet counts are done, they must be corrected for this dilution. However, platelet aggregates form more readily in dog blood collected in citrate, resulting in lower automated platelet counts than in blood collected in EDTA.485 Citrate is also the anticoagulant typically used in solutions for blood collection and storage for transfusions.371
The binding of heparin to antithrombin greatly accelerates the inhibition of thrombin by antithrombin, thereby inhibiting coagulation. FIXa, FXa, and the VIIa-TF complex also appear to be inhibited by the antithrombin-heparin complex. Heparin is used as an anticoagulant for CBCs in species where EDTA results in hemolysis. Lithium heparin is utilized as an anticoagulant when plasma (rather than serum) is used for clinical chemistry profiles. Heparin is often added to isotonic salt solutions used to flush intravenous lines and may be injected to inhibit blood coagulation in vivo. Heparin is not recommended for platelet counts because platelets tend to clump when blood is collected in heparin.281
Screening Tests For Hemostatic Disorders
Platelet Count
Cat platelets are larger than those of the other domestic animals; consequently it is not possible for impedance counters (such as the Coulter Counter S+4 and the Abbott Cell-Dyne 3500) to accurately separate cat platelets from erythrocytes by size.383 Cell counters that count platelets using laser flow cytometry (such as the Siemens Advia 120) are able to count cat platelets more accurately in whole blood. Unfortunately platelet aggregates form readily during blood collection in cats; as a result, spuriously low automated platelet counts will often be present. Platelet aggregate formation is decreased in cat blood collected in citrate, theophylline, adenosine, and dipyridamole (CTAD) vacuum tubes (BD Diagnostics, Franklin Lakes, NJ) containing platelet inhibitors.382
Platelet counts in healthy greyhounds, Polish Ogar dogs, and Cavalier King Charles spaniels are generally lower than those measured in other dog breeds. The mean platelet count in Polish Ogar dogs was 167 × 103/µL, compared with 344 × 103/µL for dogs of other breeds in Poland.345 Using an impedance counter (Abbott Cell-Dyne 3500), the reference interval (mean ± 2 standard deviations) determined for platelet counts in Greyhounds was 90,000 to 290,000/µL, compared with a reference interval of 140,000 to 380,000/µL for dogs of other breeds.448 The lower limit of the reference interval for platelet counts in Cavalier King Charles spaniels has been difficult to determine with certainty because there is a high incidence of an asymptomatic inherited thrombocytopenia with macrothrombocytes in this breed. Although overlap occurs, platelet counts below 100 × 103/µL suggest the homozygous state, with intermediate counts (about 200 × 103/µL) expected in the heterozygous state and platelet counts above 250 × 103/µL generally present in unaffected dogs.127 Platelet counts determined using impedance counters are erroneously low in Cavalier King Charles spaniels with macrothrombocytes because these counters cannot differentiate large platelets from small erythrocytes.394
Mean Platelet Volume
The mean platelet volume (MPV) is the average volume of a single platelet recorded in femtoliters (fL). Within the normal ranges of platelet counts and MPVs, there is an inverse correlation between platelet count and MPV and a direct correlation between MPV and megakaryocyte ploidy (i.e., larger megakaryocytes) in healthy humans.34,35 An inverse correlation between platelet counts and MPVs has also been reported in cats and dogs but not in horses, cattle, or goats.48
MPV determinations vary considerably with the instrument and anticoagulant used for the assay.424 Impedance cell counters can accurately determine the MPV in whole blood from dogs and horses but not in whole blood from cats. Cell counters that count and size platelets using laser flow cytometry may be able to accurately measure the MPV in whole blood of cats, but platelet aggregates often form during blood collection in cats, resulting in spuriously high MPV values.551 Of domestic animal species, cats had the highest MPVs (mean 11 fL), followed by dogs (mean 7.2 fL), horses (mean 5.0 fL), cattle (mean 4.8 fL), and goats (mean 4.2 fL) when blood was collected with sodium citrate and assayed using the same automated analyzer (Series 810, Baker Instruments Corp., Allentown, PA).48 These values would be somewhat different if EDTA were used as the anticoagulant or if a different analyzer were used for the assays.
Results from studies on the effects of anticoagulants and storage conditions on MPV are contradictory. The MPV was reported to be higher when dog blood was collected with EDTA versus citrate as the anticoagulant in an early study,217 but the MPV was reported to be lower when dog blood was collected with EDTA versus citrate as the anticoagulant in a more recent study.485 No change in MPV was found in EDTA-anticoagulated dog blood stored for a day in one study,520 but increases in MPV have been reported with storage of EDTA-anticoagulated dog blood in other studies.179,217 Discrepancies in the effect of temperature on MPV during storage have also been reported.179 These differences may be explained by the variable effects of anticoagulants on platelet activation and shape and by the various ways in which automated analyzers size platelets.424,551
A high MPV value suggests the presence of increased thrombopoiesis.112,311 Interestingly, the MPV increased in mice within 8 hours after the production of thrombocytopenia, but 40 hours were required before increased megakaryocyte ploidy was observed. The MPV has been reported to be increased in some dogs with nonimmune-mediated regenerative thrombocytopenia143 as well as in inflammatory conditions that result in enhanced platelet utilization/destruction.366,548 However, the MPV can also be high in animals with myeloid neoplasms and, in cats, with feline leukemia virus (FeLV)-induced thrombocytopenia.54,147 Cavalier King Charles spaniel dogs with inherited macrothrombocytopenia have higher MPV values because of the occurrence of a population of macrothrombocytes.127 A macrothrombocytopenia has also been reported in a pug dog with the May-Hegglin anomaly (see Fig. 5-24), as discussed in Chapter 5.164
Although the MPV is expected to be increased in response to thrombocytopenia, normal or even decreased MPVs have been associated with dogs and humans with immune-mediated thrombocytopenia (IMT), presumably because of the presence of platelet fragments (as opposed to the formation of small platelets).143,280,385 Consequently a normal MPV does not rule out enhanced thrombopoiesis, especially in primary IMT in dogs.143 In fact, most dogs with an increased reticulated platelet count do not have an increased MPV.400
MPVs have been reported to be slightly higher in hyperthyroid cats and slightly lower in hypothyroid dogs than in euthyroid animals.490 Dogs with phosphofructokinase deficiency of erythrocytes and skeletal muscle have mildly increased MPVs with normal platelet counts.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


