Chapter 26 Drugs That Modify Animal Behavior
Neurotransmitters and their Role in Abnormal Behaviors
Little is known about the cellular mechanisms of abnormal behavior in humans or animals. The most likely neurotransmitters (NTs) associated with abnormal behaviors might be identified based on the NTs targeted by drugs used to modify the behaviors. NTs identified for their role or potential role in abnormal behaviors include the biogenic amines serotonin and histamine (H1 subtype), the monoamine dopamine, the catecholamine norepinephrine, acetylcholine, gamma-aminobutyric acid (GABA), and the excitatory amino acids such as glutamate.1–3 Several of these neurotransmitters are discussed in Chapter 27.
The NT most commonly associated with abnormal behaviors is serotonin.4 Accordingly, the more effective drugs for modification of behavior are those (e.g., fluoxetine, clomipramine) that tend to be selective for serotonin. Serotonin is synthesized in the brain from tryptophan. Serotonin receptors differ in anatomic location and behavioral roles, with the majority of 5-hydroxytryptamine (5-HT) receptors being located in the gastrointestinal tract. Receptors in the central nervous system (CNS) are located in the raphe nucleus, the basal ganglia, and the limbic area. At least nine 5-HT receptor subtypes have been identified, four of which appear to be particularly important to behavior or mood.3 The 5–HT1 receptors, located primarily in the brain, are predominantly inhibitory (toward adenylyl cyclase) both presynaptically (autoreceptors; 1A and 1D) and postsynaptically (1A, 1D, 2A, 2C, and 3 and 4). Regulation of serotonin action is complex, involving both presynaptic and postsynaptic mechanisms3 with postsynaptic receptors regulating serotonin release from the presynaptic nerve endings. Serotonin may exert tonic inhibition on the dopaminergic system; drug-mediated increase may result in extrapyramidal effects.4 Badino and coworkers5 compared serotonergic and adrenergic receptor density in dogs neurologically normal except for aggressive behavior (n = 8) and normal, nonaggressive dogs (n = 8). The concentration of high affinity (serotonin) 5-HT receptors was increased in the thalamus, and low affinity 5-HT receptors were increased throughout the regions of the CNS studied (frontal cortex, thalamus, hypothalamus, hippocampus) in aggressive dogs compared with nonaggressive dogs. Riva and coworkers6 demonstrated that plasma levels of serotonin and dopamine were higher in anxious dogs (n = 22) compared with control dogs (n = 13).
In addition to serotonin, dopamine is also increased in dogs with anxiety.6 Dopamine is synthesized in neurons from l-DOPA in presynaptic vesicles. Synthesis begins with oxidation of the dietary amino acid tyrosine, followed by decarboxylation to l-DOPA.3 Those neurons in which dopamine is found lack the enzyme that converts dopamine to norepinephrine. Dopamine is removed by monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT). Dopamine receptors are distributed throughout the brain, although less so than norepinephrine receptors. Dopamine appears to be largely located in the midbrain, hypothalamus, and limbic system (the part of the brain thought to control emotions).3 Dopamine receptors (at least five subtypes) are found in portions of the extrapyramidal system responsible for coordinated movement.1,2 At least four dopamine receptors are affected by mood disorders and stereotypics; increased dopamine appears to stimulate these abnormal behaviors.1,2 Because dopamine generally suppresses acetylcholine activity, blockade of dopamine receptors generally results in an increase in acetylcholine release.4
GABA is a major inhibitory NT, being active at 30% of synapses in the human CNS, and particularly in the cortex and thalamus. Its precursor glutamate is widely distributed throughout the brain. As with norepinephrine, dopamine, and serotonin, GABA has a presynaptic transporter. Two primary receptor types, GABAA and GABAB, appear to cause postsynaptic inhibition by facilitating chloride ion influx into the neuron. Several drugs, including the benzodiazepines and barbiturates (e.g., phenobarbital), interact with the GABAA receptor in an agonistic fashion, causing neuronal inhibition.1 The physiologic and behavioral effects of GABA and its receptors have not yet been totally characterized (see Chapter 27).3
Several abnormal human behaviors have been associated with changes in excitatory NTs, including aggressive, impulsive, and schizophrenic disorders. Among the more important excitatory NTs is glutamate. Glutamate is preformed and stored in synaptic vesicles released by calcium-mediated endocytosis. Barbiturates and progesterone modulate behaviors, in part by inhibiting calcium uptake and thus release of glutamate at the NT.1,2
Acetylcholine, the most widely distributed NT in the brain,3 is preformed from choline and acetyl coenzyme A and stored at the terminal end of the synapse in vesicles. Calcium stimulates its release by exocytosis into the synaptic cleft, where it is rapidly metabolized by acetylcholinesterase. Acetylcholine generally is an excitatory NT, interacting with nicotinic and muscarinic receptors. At least five muscarinic receptors are located in the CNS. Receptors (M1) are also found in the gastrointestinal and cardiovascular systems, accounting for some of the adverse effects associated with several behavior-modifying drugs.1
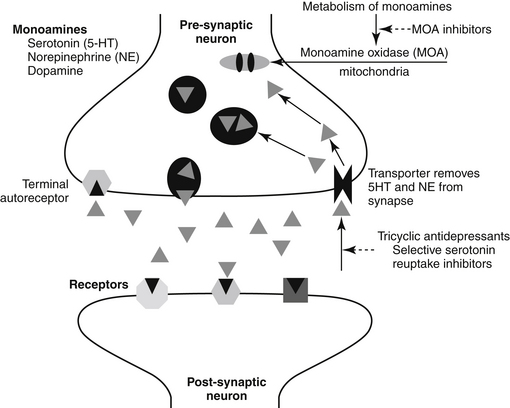
NTs tend to be formed and degraded locally. Both formation and inhibition offer pharmacologic targets. A number of drugs result in an increase in the presence of NTs in the synaptic cleft by inhibiting either the metabolism (e.g., dopamine) or the reuptake (e.g., serotonin, norepinephrine) of the NT after release (Figure 26-1).
Drugs used to Modify Behaviors
Drugs that modify behavior are classified initially on the basis of their (human) therapeutic use, followed by their chemistry and neurochemical activity.4 Included are the antipsychotic drugs (predominantly antidopaminergic in action), anxioselective drugs such as the azapirones (primarily antiserotonergic in action), drugs used to treat affective or mood disorders (antidepressants, lithium, and selected anticonvulsant drugs), and drugs used to treat anxiety and anxiety-related disorders (anxiolytics or minor tranquilizers, benzodiazepines). Other drugs include antihistamines, beta blockers, progestins, anticonvulsants, and opioid antagonists. Most of the drugs used to modify behaviors are used for treatment of other disorders, and as such may be discussed elsewhere in this volume. Only drugs that have veterinary application are discussed here. Table 26-1 provides dosing information about the use of these drugs.
Antipsychotic Drugs
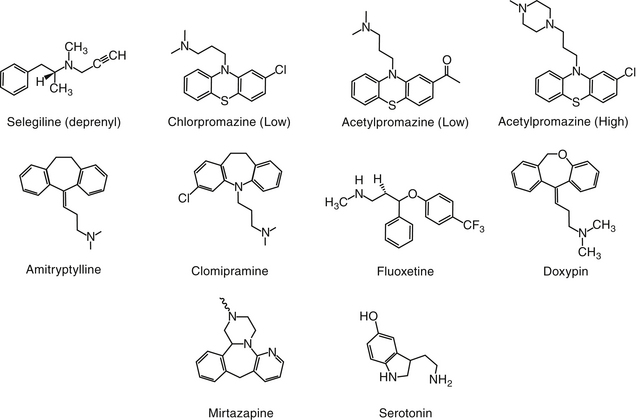
Psychotic disorders in humans involve a severe disturbance of brain function characterized by thought and speech disruption and hallucinations or delusions.3 Although psychotic disorders do not appear to occur in veterinary medicine, drugs developed for their management in humans have proved efficacious for a number of veterinary applications. Antipsychotic drugs (also called neuroleptics or major tranquilizers) are largely structurally dissimilar from one another, but the commonality among them is dopamine, and particularly D2 receptors, as a target. Drugs include the phenothiazines, the thioxanthenes (structurally related to the phenothiazines), heterocyclic dibenzepines, the butyrophenones, and diphenylbutylpiperidines (Figure 26-2).7
Structure–Activity Relationships
The largest structural class of antipsychotics is the phenothiazines or tricyclic antipsychotics, which should not be confused with the tricyclic antidepressant drugs (discussed later). The tricyclic antipsychotic drugs are represented by phenothiazine, a three-ring structure containing a sulfur and nitro group in the ring connecting two benzene rings. Substitutions on one of the benzene rings yield different drugs (e.g., chlorpromazine, promazine), which differ in efficacy (see Figure 26-2). The pharmacology also is affected by substitutions of nitro groups such that potency (but not efficacy) is reduced by an aliphatic side chain (e.g., chlorpromazine, thorazine, acepromazine, and trifluopromazine).7 The length of the side chain also determines antihistaminergic properties, with two-carbon side chains such as that occurring in promethazine being more antihistaminergic. Drugs with higher potency have a piperazine side chain, as can be found on fluphenazine and rifluoperazone. Esterification with long-chain fatty acids results in long-acting (due to slow hydrolysis and absorption) drugs (e.g., fluphenazine enanthate or decanoate).7 The use of these latter drugs in small animal veterinary medicine has yet to be established. The butyrophenone neuroleptics include haloperidol (the prototype) and droperidol. The latter is very short acting and highly sedative; thus its use is limited to anesthetic regimens.7
Pharmacologic Effects
The pharmacologic effects of antipsychotic drugs generally are similar among humans and animals.7 Phenothiazines are also categorized as tranquilizers. As tranquilizers, the phenothiazines are calming in nature, causing a decrease in spontaneous activity that generally decreases response to external stimuli.1 The predominant antipsychotic action of the phenothiazines is neuroleptic, a term derived from the effect of the drugs on human psychiatric patients and intended to contrast with signs typical of CNS depression.7 The neuroleptic effects are attributed, although not conclusively so, to the antidopaminergic effects at D2 receptors.7
Some of the neuroleptics (e.g., the phenothiazines) have high affinity for and thus also antagonize D1 dopamine receptors, although pharmacologic effects at these receptors appear to be minimal. Phenothiazines also block D3 and D4 (which are D2-like) receptors. Selected “atypical” antipsychotic drugs (e.g., clozapine) have a low affinity for D2 receptors and are not characterized by extrapyramidal effects. They are, however, characterized by α2-adrenergic antagonism. Some of the antipsychotic drugs also have affinity for serotonergic (5-HT2) receptors (e.g., clonazepam). Cholinergic and histaminergic (H1) receptors also are targeted by some of the drugs, resulting in unique pharmacologic effects among the neuroleptics. Variable interactions with different receptor types lead to unpredictable effects on the autonomic system. Among the neuroleptics, chlorpromazine has significant α-adrenergic antagonistic actions. In general, the antimuscarinic actions of neuroleptics are weak.
Neuroleptic effects include suppression of spontaneous movements or complex behaviors but minimal effects on spinal reflexes and unconditioned nociceptive avoidance behaviors. Interest in the environment is minimized (ataraxia), as are manifestations of emotion. Patients are easily aroused; ataxia or incoordination should not be evident at appropriate doses.7 Aggressive or impulsive behavior should gradually diminish. As a result, conditioned avoidance (but not unconditioned escape or avoidance) behavior and exploratory behavior are minimized. Feeding and emesis also are inhibited. At high doses, cataleptic immobility is evident (particularly in cats3), resulting in increased muscle tone (and the ability to place animals in an abnormal posture) and ptosis. Akathisia, an increase in restless activity, is an undesirable side effect that occurs in humans but apparently not in animals. Akathisia occurs as an adaptive response to increased phenothiazines in extrapyramidal tissues.7
The effects of phenothiazines occur throughout the CNS. Cortical effects are responsible for many of the neuroleptic actions. Antagonism of D2 receptors is largely responsible for the various extrapyramidal effects of the drugs. Many of these sites appear to be spared from the adaptive changes of tolerance.7 Neuroleptics have been associated with an increased incidence of seizures.8 Selected drugs lower seizure threshold as well as induce discharges typical of epileptic seizures. Aliphatic, low-potency phenothiazines are particularly characterized by this effect.7 Although the effect is more likely to occur in patients who are epileptic or who are predisposed to seizures, the effect is also a dose-dependent characteristic of some drugs. These drugs are contraindicated in human epileptic patients or patients undergoing withdrawal from central depressants.7 The relevance of this precaution to dogs is not clear. Both Tobias and coworkers9 and McConnell and coworkers10 retrospectively studied the frequency of seizures in epileptic dogs treated with low doses of acepromazine and found no increased risk (see also Chapter 27). Indeed, acepromazine was associated with decreased seizure activity in some of the patients that did not respond to other therapies. However, because the author experienced induction of seizures in a non-epileptic patient (lead poisoning) immediately after intravenous administration of acepromazine, caution is warranted when using phenothiazine derivatives in epileptic patients. Increasing doses slowly and accompanying with anticonvulsant therapy are indicated if the drugs must be used by epileptic patients.
The neuroleptic drugs have a number of effects in the limbic system. Although D2 antagonism occurs in the limbic system, because D3 receptor stimulation may be responsible for many of the behaviors targeted by neuroleptics, attempts are being made to identify D3-selective drugs for treatment of psychoses.7 Neuroleptics stimulate prolactin secretion in human beings. Indeed, the potency of neuroleptic action and ability to cause prolactin secretion are well correlated for most drugs. Pageat and coworkers11 demonstrated that prolactemia correlates with response to selected behavior-modifying drugs. Tolerance is not likely to develop to this side effect. In humans prolactin secretion caused by neuroleptics also is responsible for breast engorgement and galactorrhea. Releases of growth hormone and corticotropin-releasing hormone occur in response to stress; neuroleptics (especially chlorpromazine) also interfere with the release of growth hormone, although apparently not sufficiently for treatment of acromegaly. Impaired release of serotonin may result in weight gain (particularly with low-potency drugs), and impaired glucose tolerance and insulin release may be impaired in “prediabetic” patients (especially with chlorpromazine).7
In the brainstem the neuroleptics have little effect, even in cases of acute overdosing. Life-threatening coma is rare. In contrast, most neuroleptics protect against nausea and emesis at the chemoreceptor trigger zone in the medulla. These effects occur at low doses. Potent piperazines and butyrophenones are also often effective against nausea stimulated by the vestibular system.7
Phenothiazines characterized by lower potency have a predominant sedative effect that is more apparent initially but tends to decline as tolerance develops. The phenothiazines are characterized by anxiolytic effects, but more specific anxiolytic drugs are available. In addition, the risk of either autonomic (e.g., low-potency drugs) or extrapyramidal (e.g., highly potent drugs) effects increases the likelihood of causing anxiety.7
Disposition
The antipsychotics are characterized by variable bioavailability, high lipophilicity, high protein binding, and accumulation in a number of tissues. Elimination occurs primarily through hepatic metabolism. In humans the elimination half-life is long, ranging from 20 to 40 hours. Biologic effects persist for more than 24 hours, allowing once-daily therapy in humans.7 Metabolites can be detected in urine for several months.
Side Effects and Toxicity
The antipsychotic drugs tend to be very safe; lethal ingestion is rare in human patients. Side effects tend to reflect the pharmacologic actions of the drugs, including effects of the CNS and cardiovascular, endocrine, and autonomic systems.7 In human patients other effects include dry mouth, blurry vision, and constipation. Urinary retention may occur in male patients with prostatitis. At high doses antipsychotics may cause catalepsy, characterized by immobility, increased muscle tone, and abnormal postures.4 Extrapyramidal neurologic side effects reflecting inhibitionof dopamine occur in humans but have not been reported in animals. However, involuntary motor movements associated with extrapyramidal effects may be confused with seizures. The risk of seizures may be increased, but risk factors are not known (discussed previously). Some animals exhibit signs of hyperactivity after treatment with acepromazine.3 In addition, at least one report cites increased agitation and irritability after treatment of aggression with acepromazine.12 Jaundice has occurred in humans after taking chlorpromazine and may resolve despite continued treatment. Blood dyscrasias, including leukopenia, eosinophilia, and leukocytosis, occur but are less common with low-potency phenothiazines. Skin reactions tend to be common in people, again more commonly with low-potency phenothiazines. Small doses of acepromazine (0.1 mg/kg, given subcutaneously 30 minutes before testing) appear to have no effect on intravenous glucose tolerance testing in dogs.13 Acepromazine sedation (0.03 mg/kg given intramuscularly 30 minutes before measurement) markedly increased peak latencies but minimally affected peak amplitude of middle-latency auditory-evoked potential in dogs (n = 12).7,14
Drug Interactions
Chlorpromazine is used in combination anesthetic regimens because of its ability to potentiate central depressants. Effects of analgesics and sedatives also can be enhanced. Interactions with antihypertensive drugs can be unpredictable and are more likely to be adverse with low-potency products.7 Selected phenothiazines can antagonize the positive inotropic effects of digoxin.
Clinical Indications
In general, the use of phenothiazines for treatment of aggressive behavioral abnormalities is inappropriate because both normal and abnormal behaviors are blunted. Acepromazine is particularly problematic. Restraint of aggressive dogs with the drug renders dogs more likely to be reactive to noises and more easily startled.1 In addition, because the degree and duration of tranquilization vary, reactions in dogs are unpredictable. Phenothiazines are not selective as antianxiety drugs but can reduce responsiveness in general, and thus they are useful in some cases of episodic anxiety.3 Thioridazine has been used in one case of aberrant motor behavior.15 Newer atypical antipsychotics (e.g., risperidone) may prove useful for treatment of selected abnormal behaviors (e.g., environmental-specific anxieties, impulsive explosive disorders).4
Antidepressant Drugs
Much of the information regarding the use of these mood-modifying drugs in animals has been extrapolated from human use. These drugs are characterized by clinical pharmacology and mechanisms of action that are likely to differ markedly among animals. Yet limited scientific information is available to guide their use in animals. Among the human mood-modifying drugs that are approved for their use for treatment of behavioral disorders in either dogs or cats are the tricyclic antidepressants (TCAs) (clomipramine), the MAO inhibitors (selegiline), and selective serotonin reuptake inhibitors (SSRIs; e.g., fluoxetine). Affective (behavior) disorders targeted by antidepressants in humans range from depression to manic–depressive disorders. In animals the list of targeted disorders is much greater and may be publically perceived to include any type of behavior deemed unacceptable by pet owners.
Tricyclic Antidepressants
Structure–Activity Relationships
The TCAs are among the most frequently prescribed drugs in human behavior medicine. Their name reflects their chemical structure (see Figure 26-2). The TCAs were identified as a group of potentially useful drugs for the modification of behavior in the 1940s after the generation of a number of drugs with antihistaminergic, sedative, analgesic, and antiparkinsonian effects. Imipramine was selected on the basis of its hypnotic and sedative effects. Imipramine differs from phenothiazine only by the replacement of sulfur with an ethylene bridge, yielding a seven-member ring. This compound proved ineffective in quieting agitated psychotic patients but very effective for selected mood disorders.7
The search for chemically related compounds yielded a number of additional drugs. Clomipramine, amitriptyline, and doxepin are all derivatives of imipramine.7 Each contains a tertiary amine at one of the substitution sites on the seven-member ring. Desipramine, a major metabolite of imipramine, and nortriptyline, the N-demethylated metabolite of amitriptyline, are secondary amine tricyclics. Protriptyline and trimipramine are other TCAs with few veterinary applications.3 Differences in the effects of NT reuptake vary with the different amine structures.7
Mechanism of Action
The mechanism of action of the TCAs (and MAO inhibitors and SSRIs) is blockade of the mechanisms of physiologic inactivation (see Figure 26-1). For the TCAs the mechanism is inhibition of reuptake at presynaptic biogenic amine NT receptors in the brain. As reuptake is inhibited, the concentration of NTs increases, prolonging their actions (CNS stimulation). Differences in NTs affected reflect, in part, the chemical structures.7 Imipramine and its derivatives with a tertiary amine side chain block norepinephrine reuptake but have little effect on dopamine reuptake.
The secondary amine derivatives of imipramine are potent and highly selective inhibitors of norepinephrine reuptake; however, they are characterized by fewer autonomic and anticholinergic effects. Tertiary amines that are metabolized in the patient to secondary amines will have effects on norepinephrine reuptake. Clomipramine has marked effects on serotonin reuptake and minimal effects on other NTs or receptors, although a poorly understood dopaminergic effect has been described. Its active metabolite appears to inhibit norepinephrine uptake as well.16 Doxepin is characterized by greater antihistaminergic actions (thus explaining its frequent recommendation for chronic pruritus). Although amitriptyline is the most commonly prescribed drug for animals, clomipramine has recently been approved for treatment of separation anxiety in dogs.
The effect of clomipramine (3 mg/kg once daily orally for 6 weeks) on the concentration of monoamines (5HIAA [5-hydroxyindoleacetic acid], HVI [homovanillic acid] and MHPG [methoxy4-hydroxyphenylglycol]) in the cerebrospinal fluid was compared with that of placebo using a randomized crossover design in otherwise behaviorally normal dogs (n = 6) with obsessive–compulsive disorders (OCDs).16 The concentration of monoamines did not significantly differ (power of the study now known), leading the authors to conclude that clomipramine did not affect monoamine turnover in the brains of behaviorally normal dogs, further confirming the need for studies in affected dogs.
Pharmacologic Effects
Adaptation to pharmacologic effects
The effects of TCAs at presynaptic and postsynaptic receptors and autoregulation result in complex responses that are not well understood. Although inhibition of reuptake occurs very rapidly, peak effects still take several weeks. This prolonged time to maximal effect reflects in part disposition (e.g., time to steady-state for the parent compound and active metabolites; see later discussion of clinical pharmacology) but also appears to reflect adaptation in the CNS to changes in NT concentrations at the synapse. Administration of a TCA results in an immediate decrease in the synthesis and release of norepinephrine or serotonin (depending on the major target of the TCA) in selected areas of the brain. The effects appear to be mediated presynaptically through autoreceptors (α2 or serotonin, respectively). Turnover, however, gradually normalizes within 1 to 3 weeks. Autoreceptors appear to be downregulated and become desensitized to the presence of the TCA.7 The number and sensitivity of postsynaptic adrenergic receptors do not appear to be affected by continued use of a TCA.
Adaptive responses appear to influence the pharmacologic properties of TCAs at adrenergic receptor sites. The TCAs have a moderate affinity for α1-receptors and only limited affinity for α2-receptors and β-receptors. Changes in serotonin receptors after repeated treatment with TCAs are complex. Although the impact is not clear, the general effect appears to be increased sensitivity to serotonin. This may be an important component to the outcome of prolonged treatment.7 The TCAs also appear to influence the effect of other NTs and their receptors, including GABA (unknown significance), and dopamine (D2; desensitization of autoreceptors resulting in mood elevation). Other factors to consider regarding adaptation to TCA effects include changes in adenosine monophosphate–dependent protein kinases and potential changes at the level of gene expression.7
In addition to tolerance (to sedative and autonomic effects), physical dependence on the TCA can develop. Physical dependence after acute withdrawal is manifested in human patients as malaise, chills, coryza (head cold), and muscle aches.7 Slow discontinuation of the drug is recommended.
Autonomic nervous system
The predominant effect of TCAs on the autonomic nervous system appears to reflect inhibition of norepinephrine transport into adrenergic nerve terminals and antagonism of muscarinic cholinergic and α1-adrenergic responses to the NTs. Blurred vision, dry mouth, constipation, and urinary retention at therapeutic doses (documented in humans) appear to reflect anticholinergic effects.7
Cardiovascular system
Cardiovascular effects of TCAs occur at therapeutic doses and can become life threatening with overdose. Postural hypotension occurs in humans (unlikely in dogs or cats) as a result of α–adrenergic blockade. Mild sinus tachycardia results from inhibition of norepinephrine uptake and muscarinic (M1) blockade.7 Conduction time is prolonged, especially at concentrations above 200 ng/mL. The TCA can also directly suppress the myocardium.7 The myocardial depressant effects are greater in the presence of underlying cardiac disease.
Clinical Pharmacology
The disposition of the TCA favors adverse reactions in that the characteristics of disposition are those that tend to vary greatly among animals, and extrapolation between species is complicated. Unfortunately, the disposition has not been scientifically studied in animals except for clomipramine, and information for other drugs or species is extrapolated from human or canine data. The TCAs are very lipophilic. As such, they are well absorbed after oral administration. They can, however, undergo marked first-pass metabolism. Typical drug concentrations achieved in humans at recommended doses are 100 to 250 ng/mL for amitriptyline, 150 to 500 ng/mL for clomipramine, and 200 to 300 ng/mL for imipramine.7 High doses can cause anticholinergic effects on the gastrointestinal tract, slowing absorption or making it erratic. Absorption in humans can result in peak concentrations as rapidly as 2 hours or as long as 12 hours after administration.7 The drugs are very highly protein bound, but unbound drug is characterized by a very large volume of distribution (10 to 15 L/kg in human patients), contributing to a long elimination half-life of the parent drug or its metabolites. Drug may bind avidly to selected tissues.
Drugs are eliminated by hepatic (oxidative) metabolism with the main reaction being N-demethylation to the active metabolite, desmethylclomipramine (DCMP). This reaction is catalyzed by CYP3A4, CYP2C19, CYP2C9, and CYP1A2. The parent and metabolite are hydroxylated by CYP2D6 and then glucuronidated.17,18 Metabolism is variable among human and canine patients, accounting for plasma concentrations that differ by tenfold to thirtyfold. Species and gender differences in metabolism or pharmacokinetics have been demonstrated for clompramine.17,18 Metabolism of TCA yields active and inactive metabolites. It is not clear what percentage of the antidepressant activity of the TCA is associated with the metabolites. Metabolites generally have an elimination half-life that is twice or more that of the parent compound.7 Thus accumulation of the metabolites can result in a marked proportion of the pharmacologic effect of TCAs.
Clomipramine is approved for use in dogs for treatment of separation anxiety. After multiple oral administration in dogs, both clomipramine and its principal active metabolite, desmethylclomipramine, accumulate in a dose-dependent manner at doses ranging from 1 to 4 mg/kg.19 Food does not appear to alter the rate or the extent of oral absorption,20 with bioavailability of clomipramine being 20% and of the metabolite 140%. The ratio of metabolite to parent in dogs approximates 0.6, varying across time.21 After a single dose of 3 mg/kg in normal dogs (n = 6), clomipramine Cmax was quite variable for both clomipramine (16 to 310 ng/mL at 0.75 to 3.1 hr) and desmethylclomipramine (21 to 134 ng/mL at 1.4 to 8.8 hr).22 After multiple dosing, (28 d) clomipramine Cmax again was markedly variable at 43 to 222 ng/mL (at 3 to 8 hr), compared with 21 to 134 ng/mL at 1.4 to 8.8 hr, for desmethylclomipramine. Hewson and coworkers21 reported trough concentrations of clomipramine ranging from 2 to 17 ng/mL and desmethylclomipramine ranging from 2 to 15 ng/mL following 26 days of dosing at 3 mg/kg; neither compound was detectable in three of six dogs at trough times (24 hr). Protein binding of both the parent compound and its metabolite are greater than 96%. Hepatic clearance of clomipramine in dogs is flow-limited; changes in hepatic blood flow, but not hepatic mass (i.e., CYP activity), should be expected to alter clomipramine elimination. However, rapid clearance is balanced by a very large volume of distribution. Despite this large volume, clomipramine elimination half-life is shorter and markedly variable in dogs ranging from 1.2 to 16 hours after a single oral dose in dogs (n = 6) compared to 20 in humans. After multiple (28 days) dosing, both variability and duration of half-life decreased to 1.5 to 9 hours.21 An average half-life of 5 to 6 hours was reported for dogs in a subsequent study.19 Half-life of the desmethyl clomipramine in dogs also is short (2 to 3.8 hours, compared to 36 hours in humans),19,21 with total concentration of the metabolite being less than that of the parent compound. Steady-state concentrations are reached by 4 days or less, resulting in response within 1 to 2 weeks. Cats metabolize (based on in vitro hepatic microsomal studies) clomipramine more slowly than do dog or rats, with male cats being less efficient metabolizers.17 The pharmacokinetics of clomipramine and its major metabolite desmethylclomipramine have been described in cats (n = 6) using a randomized crossover single-dose oral (0.5 mg/kg) and intravenous (0.25 mg/kg) administration study).23 After oral administration, Cmax (μg/mL) was 0.87 ng/mL at 6.2 hours. After intravenous administration, volume of distribution and clearance were 0.393 L/kg and 0.833 mL ∗ min/kg, respectively, resulting in an elimination half-life of 12.3 hours. Area under the curve (AUC) of clomipramine and desmethylclomipramine after oral administration were 948 and 613 ng ∗ min/mL, respectively. Oral bioavailability of clomipramine and its active metabolite approximated 30%. Variability among cats was marked. On the basis of these data, Lainesse and coworkers18 found no correlation between either clomipramine or desmethylclomipramine concentrations and sleep scores. However, gender differences in metabolism may have contributed to the lack of correlation.
Of the TCAs, only amitriptyline appears to have been studied after transdermal delivery in cats. Plasma drug concentrations were not detectable in cats (n = 6) receiving a single 5-mg transdermal (pluronic lecithin organo [PLO]) gel dose. This compared with a mean peak concentration of 61 ± 6.2 ng/mL after oral administration of 5 mg.24 The author has demonstrated, using a randomized crossover, dummy-placebo controlled design, that although amitryptylline administered as a transdermal gel achieved detectable concentrations in 5/6 cats receiving 2 mg/kg twice daily for 3 weeks, bioavailability was very poor. After 3 weeks of dosing, serum drug concentrations were 46 to 101 ng/mL after gel administration compared to 1336 to 5230 ng/mL after oral administration at the same dose. The disappearance half-life in the cats receiving the oral drug (based on peak and trough dosing) was markedly variable among cats, ranging from 20 to approximately 85 hrs after 3 weeks. Two of the cats receiving the gel developed marked aural erythema and scaling. Despite the very high concentrations, 4/5 cats tolerated the oral administration well; the one cat with concentrations of 5230 ng/mL was depressed, with resolution occurring once the drug was discontinued. Interestingly, disappearance half-life of amitryptyilline was only 20 hrs in this cat, suggesting that oral bioavailabilitymay have been contributing to the high serum concentrations.
Side effects
Up to 5% of human patients receiving a TCA react adversely. Sedation is common with the TCAs.3 The most common reactions reflect antimuscarinic effect or overdosing. Cardiac toxicity (reflecting a quinidine-like membrane-stabilizing effect) is a less frequently reported but serious effect, but not apparently with clomipramine administration in dogs. The effect may be lethal at 15 mg/kg.4 Tachycardia may also reflect anticholinergic effects and must be differentiated from true cardiotoxicity. Other side effects include dry mouth, gastric distress, constipation, dizziness, tachycardia, or other arrhythmias, blurred vision, and urinary retention (particularly problematic in the presence of prostatic hypertrophy).7 Weakness and fatigue reflect CNS effects. Cardiac toxicity is more likely in patients who start therapy with cardiac disease. In healthy patients the most likely cardiac response is hypotension as a result of α-adrenergic blockade.
An undesirable side effect of antidepressant drugs in humans is referred to as the “switch process.” Patients undergo a transition from depression to hypomanic or manic excitement.7 This effect has not been reported in animals. Confusion and delirium are behavior aberrations that occur commonly in human patients, with the incidence of 10% in all patients, increasing to more than 30% in elderly patients over age 50 years.7 Miscellaneous toxic effects in human patients include leukopenia, jaundice, and skin rashes. Weight gain occurs, particularly with those drugs that are selective for serotonin reuptake. Isolated reports that address the side effects of TCAs in dogs are uncommon. Goldberger and Rapoport25 reported side effects in 5 of 13 dogs receiving clomipramine for lick granuloma. Clinical signs included lethargy, anorexia, diarrhea, and growling.
Acute poisoning with TCAs is common in human patients (accidental or intentional) and appears to be a significant potential problem in animals.26 Symptoms in humans vary and are complex. Excitement and restlessness may be accompanied by myoclonus or tonic–clonic seizures. Coma may rapidly develop, associated with depressed expiration, hypoxia, hypothermia, and hypotension.7 Anticholinergic effects include mydriasis, dry mucosa, absent bowel sounds, urinary retention, and cardiac arrhythmias, including tachycardia. Clinical signs reported after accidental ingestion in animals26 include hyperexcitement and vomiting as early manifestations, followed by ataxia, lethargy, and muscular tremors. Bradycardia and other cardiac arrhythmias occur later. These later signs occurred shortly before death in experimental animal models of TCA toxicosis.26
Treatment for TCA toxicosis is supportive, including respiratory (intubation) and cardiovascular support. Gastric lavage with activated charcoal can be used early. Emetics probably should be avoided because of the risk of aspiration pneumonia in seizuring animals (some antiemetics may further predispose the animal to seizures). Short-acting barbiturates (or similar drugs) without pre-administration of atropine are preferred for anesthetic control during gastric lavage. Cathartics (sorbitol or sodium sulfate–Glauber’s salt) can be of benefit. Magnesium sulfate should not be used because impaired gastrointestinal motility can facilitate absorption of magnesium. Resolution of coma may require several days; the threat of cardiac arrhythmias likewise persists for several days. Pharmacologic interventions for cardiac arrhythmias have not been well established. Alkalinization (sodium bicarbonate sufficient to maintain blood pH above 7.5: 2 to 3 mEq/kg, administered intravenously over 15 to 30 minutes) may prevent death by increasing protein binding and increasing cardiac automaticity (as a result of potassium shifts).26 Traditional cardiac drugs, including antiarrhythmics and digoxin, are contraindicated in human patients. However, phenytoin may provide safe antiarrhythmic effects and in human patients is useful for treatment of seizures.7 Relevance to dogs or cats is not clear: phenytoin has a short half-life in dogs and is associated with adverse events when administered IV (see Chapter 27). Diazepam is indicated for acute management of seizures. β–adrenergic receptor antagonists and lidocaine may be useful.7 The risk of tonic–clonic seizures is increased in human patients, particularly at high doses.
Clomipramine generally is associated with less sedation than with the other TCAs.27 Systemic phospholipidosis (excessive accumulation of phospholipids in tissues) occurs with chronic administration of cationic, amphophilic drugs, including the TCAs, and fluoxetine.
Indications
The TCAs have been recommended among animal behaviorists for most abnormal behaviors manifested in dogs or cats. These include, but are not limited to, behaviors associated with fear and aggression,27,28 stereotypics, OCDs or self-mutilation disorders, and excessive barking. The use of clomipramine is discussed later with specific disorders. However, using an open trial, Seksel and Lindeman29 reported the efficacy and tolerance of clomipramine (1 to 2 mg/kg every 12 hours for the initial dose, increased up to 4 mg/kg twice daily) in a variety of behavioral disorders in dogs (n = 24). Included in the study were OCD (n = 9), separation anxiety (n = 14), and noise phobia (n = 4). Treatment included behavior modification and continued for at least 1 month after resolution of clinical signs. Resolution to marked improvement of clinical signs was reported in 67% of the dogs, whereas 21% reported slight to moderate improvement versus no change in 12.5% (3 of 24) animals. Clomipramine discontinuation (without return of the abnormal behavior) was described as successful in five of the nine animals in which it was attempted.
Contraindications
The TCAs are not recommended for animals with a metabolic disease. Specific contraindications include a history of cardiac or hepatic disease, seizures, glaucoma, hyperthyroidism, and thyroid hormone supplementation.27,28
Drug Interactions
The TCAs can interact with a number of other drugs. Competition for protein-binding sites with other highly protein-bound drugs can result in increased drug concentrations; however, this may be balanced by increased hepatic clearance. Drugs that affect drug-metabolizing enzymes, through either inhibition or induction, also affect the clearance of TCAs. The sequelae of the impact are difficult to predict because active metabolites similarly are affected. In general, however, drugs that inhibit metabolism are likely to result in greater drug accumulation and increased risk of toxicity. TCAs and other antidepressants also can compete with other compounds for metabolism. The drugs themselves may affect metabolism of other drugs. Clomipramine inhibits the metabolism of other drugs; but a more relevant interaction may be the impact of other drugs on the metabolism of clomipramine (e.g., cimetidine).7 The antidepressants potentiate the effects of sedative drugs. The TCAs should not be used in combination with other drugs that modify CNS NTs. Included are MAO inhibitors and amitraz.27,28 In human patients a potentially lethal interaction has been reported when a TCA, particularly one that inhibits serotonin uptake, is combined with an MAO.7 The term serotonin syndrome has been applied to this interaction, which is characterized by restlessness, muscle twitches, hyperreflexia, shivering, and tremors.30 Drugs or herbal products whose combination might result in the serotonin syndrome are indicated in Table 26-2.
Table 26-2 Drugs or Supplements Whose Combined Use Increases the Risk of Serotonin Syndrome
| Drug Class | Interacting Drug Class | Example Drugs |
|---|---|---|
| Monamine oxidase inhibitors | Tricyclic antidepressants | Amitriptyline |
| Imipramine | ||
| Clomipramine | ||
| Miscellaneous | Isocarboxazid | |
| Phenelzine | ||
| Serotonin uptake inhibitors | SSRI | Fluoxetine |
| Fluvoxamine | ||
| Paroxetine | ||
| Sertraline | ||
| Citalopram | ||
| Serotonin and norepinephrine uptake inhibitors | Trazazone | |
| Bupriopion | Venlaflaxine | |
| Opioids | Fentanyl | |
| Mepiridine | ||
| Tramadol | ||
| Dextromethorphan | ||
| Antimicrobials | Antibacterials | Linesolid |
| Antivirals | Ritonavir | |
| Antiemetics | Metoclopramide | |
| Ondansetron | ||
| Granisetron | ||
| Herbals | St. John’s wort | |
| SAMe | ||
| Ginseng | ||
| Substances of abuse | MDMA | |
| Cocaine |
Clinical Use
Most drugs take 2 to 3 weeks for clinical efficacy to be realized. The exception might be amitriptyline, which may cause a response in 3 to 5 days; however, maximum response may take longer, particularly in cats, for which the half-life may be long.27 Dogs can respond to clomipramine in 1 to 2 weeks. In an approval study, clomipramine (Clomicalm) treatment with behavior modification improved separation anxiety in dogs compared with behavior modification alone (according to package insert). King and coworkers31,32 (manufacturer sponsored studies) studied the efficacy of clomipramine for treatment of separation anxiety in dogs. Using a parallel, randomized, double-blinded design, researchers gave dogs with “hyper attachment” behavior (n = 97) either a low dose (0.5 to 1 mg/kg twice daily; n = 25), a standard dose (1 to <2 mg/kg twice daily; n = 28), or no dose of clomipramine (n = 32) along with behavior therapy for 12 weeks. At each of three assessment periods, treatment dogs responded better, and overall, to therapies three times more rapidly than nontreatment dogs in all behaviors (destructive or inappropriate elimination) with the exception of vocalization. Among the treatment dogs was an epileptic; this dog had no seizure episodes during the treatment period. Differences in the frequency of side effects among treatment groups were vomiting and gastritis, which occurred in 13/25 and 8/28 dogs in the low and standard group, respectively, compared with three in the placebo group, and lethargy or sleepiness in four of the standard group compared with two in the other two groups. Interestingly, one healthy dog collapsed with hyperthermia (111°F) and disseminated intravascular coagulopathy. No other underlying illness was reported; the dog survived the episode, but clomipramine was discontinued. In their follow-up questionnaire-based study,32 12 dogs continued to receive clomipramine long term (13 to 16 months or more), with 10 of these dogs improving further. No increases in adverse events were identified. Behavior changes were also studied in dogs for which the drug was either discontinued (n = 48) or had not been administered within 4 months of the questionnaire (n = 16). However, the data are confounded by the use of other behavior-modifying drugs. Behaviors worsened in the first 2 weeks after therapy was discontinued in three of ten dogs receiving low-dose clomipramine; however, no dogs in the standard-dose group exhibited clinical signs potentially consistent with withdrawal. For treatment groups, after 2 weeks behavior worsened in 13% to 15% of dogs compared with 23% of dogs receiving placebo. Six of nine dogs for which clinical signs worsened had not responded totally to therapy during the trial, whereas three of nine had been considered successes. The time to worsening was longest for the standard dose group. On the basis of response to clomipramine, the authors raised the possibility that treatment accompanied by behavioral therapy might allow permanent control.
Clomipramine has been studied for acute management of anxiety in dogs. Frank and coworkers33 prospectively studied transport-induced stress in Beagles treated with clomipramine (2 mg/kg twice daily for 7 days). Compared with placebo, treatment only tended to reduce clinical signs of panting or drooling; however, cortisol concentrations increased less in treated versus untreated controls after three 1-hour trips.
Therapeutic drug monitoring may facilitate the safe and effective use of the drugs. In human patients plasma concentrations that range between 100 and 250 ng/mL are most likely to cause satisfactory antidepressant effects; toxicity can be expected at concentrations above 500 ng/mL, with fatal consequences likely as concentrations approach 1000 ng/mL.7 Variability among human patients (and presumably among animals) supports the use of monitoring to guide therapy. Monitoring to avoid toxicity is, however, complicated by the recognition that serum concentrations by themselves are not reliable predictors of toxic responses. Because of the risk of withdrawal associated with physical dependence, discontinuation of TCAs should occur over a week or longer if therapy has been prolonged.7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


