Chapter 17 Drugs Affecting Urine Formation
Renal Physiology and Drug Therapy
Extracellular Fluid
The physiology of body fluids and the role of the kidney are also addressed in Chapters 16 and 18 The volume of extracellular fluid (ECF) is determined primarily by total body sodium content for two reasons: First, sodium is the major constituent of ECF; secondly, active transport mechanisms control sodium in intracellular and extracellular compartments.1 Control of ECF involves cardiovascular, renal, and central nervous systems. Because these systems are closely integrated, one system can compensate for the failure of another system, thus ensuring that salt and water excretion remain appropriate even with changes in blood pressure. Among the compensatory mechanisms is autoregulation of renal blood flow through efferent arteriolar resistance. Because regulatory mechanisms adjust both short- and long-term sodium and water transport rates, small increases in mean arterial blood pressure can cause a marked increase in sodium excretion, ultimately changing total body sodium.1 However, a check-and-balance system exists whereby if sodium balance becomes negative, as sodium concentration in the ECF decreases, causing thirst and water intake to decrease accordingly until ECF sodium concentration normalizes. A positive balance does the opposite, resulting in an increase in water intake. These integrated systems maintain a sodium and water excretion that equals that of intake minus any lost through nonrenal mechanisms (e.g., feces, sweat).
Renal Transport of Fluids and Electrolytes
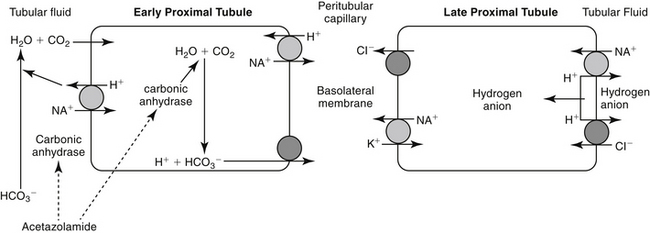
The proximal tubules are responsible for 66% of the sodium and glomerular filtrate reabsorbed by the kidney. The primary pathway of fluid and electrolyte reabsorption in the renal tubule begins in the lumen and progresses through the cell and the interstitial fluid and into the capillary. Two water-permeable cell membranes are traversed during reabsorption. In the proximal tubule, active sodium ion reabsorption from the lumen into the cell generates an osmotic gradient in the lumen that leads to an almost simultaneous movement of water into the cell. Sodium reabsorption that begins with entry across the luminal membrane continues with movement across the basolateral membrane into the interstitial space (Figure 17-1). Basolateral movement is the energy-dependent process, fueled by a Na/K-dependent ATPase located on the basolateral membrane. The exchange rate of two K+ for each three Na+ entering the interstitial fluid provides an electrochemical gradient that favors passive entry of Na+ from the lumen into the cell. The concentration gradient generated by the basolateral movement determines the rate of sodium movement from the lumen. Chloride (and to a lesser degree, other anions) follows sodium, maintaining electroneutrality of the reabsorbate and a slight electronegativity of the cell compared with the luminal contents. The concentration gradient also favors passive movement of potassium from the interstitium into the cell, where it is used to continue the Na-K exchange. Although movement of sodium from the lumen into the cell is passive (albeit at the cost of an active basolateral efflux), sodium reabsorption into the cell is facilitated by three additional entry mechanisms: diffusion with chloride (quantitatively the most important), co-transport with uncharged or acidic anions, and countertransport with hydrogen ions (important to acid–base regulation and the site of carbonic anhydrase action). Although transport of sodium, water, and other electrolytes raises interstitial pressure, movement into peritubular capillaries facilitates continued reabsorption. Bicarbonate processing also occurs in the proximal tubule.
Unlike the proximal tubule, cells of the descending limb of the loop of Henle do not appear to be equipped with specialized transporting systems. The cells are relatively impermeable to sodium, chloride, and potassium but are permeable to water. Water moves from the lumen into the interstitium, causing electrolyte concentrations to progressively increase in the lumen until a maximum is reached at the bend. Diuretic drugs do not appear to be active in the descending portion of the tubule. In the ascending limb of the loop, chloride is transported “uphill,” achieving intracellular concentrations that are higher than predicted (based on the Nernst equation).2 Chloride movement occurs by a Na (downhill), K+-2Cl– (uphill) co-transport system, with Na+, K+-ATPase in the peritubular membrane providing the energy source. High intracellular chloride concentration facilitates chloride movement into the interstitium. The ion transports in the ascending loop of Henle are critical for proper function of the countercurrent mechanism in the renal medulla. The ascending loop of Henle tubule is not permeable to water, and fluid in the lumen becomes progressively diluted (Figure 17-2). About 25% of sodium-chloride reabsorption and 40% of potassium reabsorption occurs in the ascending loop of Henle, although only about 15% of the filtrate is reabsorbed in this region.2 In contrast to the proximal tubules, Na+-H+ exchange does not appear to occur in the loop of Henle, and little if any bicarbonate is processed.
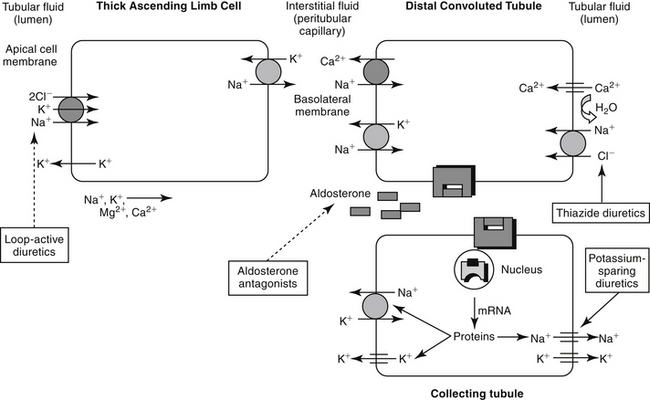
Reabsorption of water and electrolytes in the distal tubule and collecting ducts is variable. Sodium and chloride are reabsorbed against a concentration gradient. Because the early distal tubule is impermeable to water, an unfavorable concentration gradient is generated that limits the effectiveness of a sodium pump. Sodium and potassium contents in the urine are closely regulated by an aldosterone-sensitive mechanism in the distal late tubule (see Figure 17-2). Aldosterone signals the synthesis of a protein that increases the sodium and potassium permeability of the luminal membrane. As sodium moves in, potassium simultaneously moves from the interstitium into the cell. Electrogenic movement of sodium into the cell causes an electronegativity in the lumen that attracts potassium from the cell. Whether potassium is reabsorbed or excreted is determined primarily by plasma potassium, which in turn tends to depend on dietary intake.2
In the medullary collecting ducts, only small amounts of sodium chloride and potassium are reabsorbed. Antidiuretic hormone increases permeability of the cell membrane to water, which moves from the lumen, following the previously established medullary osmotic gradient. Less than 5% of filtered sodium is reabsorbed in the distal tubule and medullary collecting system under normal conditions.2
Atrial Natriuretic Hormone
Atrial natriuretic hormone (ANH), a “natriuretic factor” first described in 1984, is involved in the control of ECF. It is synthesized from a prohormone and stored as a peptide in granules in atrial myocardial cells. Concentrations increase above baseline when either the ECF expands, blood pressure increases, or dietary salt intake increases. Renal blood flow and glomerular filtration are subsequently increased. Sodium excretion also increases, presumably by a direct tubular action. Peripheral vasodilation can result in decreased blood pressure. Effects occur rapidly but are not sustained, suggesting that ANH is a mechanism that can restore equilibrium rapidly.2
Diuretic Therapy
Therapeutic Use of Diuretics
Three strategies exist for movement of inappropriate fluid accumulation (edema): correction of the underlying disease (often not possible), restriction of dietary or other sodium intake, and administration of diuretics. Of these, diuretics remain the cornerstone for treatment of edema or volume overload, particularly that which is life threatening.1 Although diuretics increase the rate of urine formation, their therapeutic indications include maintenance of urine flow; mobilization and reduction of inappropriate ECF stores, such as that manifested as edema or ascites; correction of specific ion imbalances; reduction in the rate of intraocular fluid formation; reduction of blood pressure; and reduction of pulmonary capillary wedge pressure.2
Targets of Diuretic Therapy
Diuretics are classified by their mechanism of action and include the loop diuretics, carbonic anhydrase inhibitors, thiazides, osmotic diuretics, and potassium-sparing diuretics (Figure 17-3, Table 17-1). Rational selection of diuretics relies on an appreciation of their mechanisms of action. With the exception of the osmotic diuretics and carbonic anhydrase inhibitors (the latter targets sodium bicarbonate), each class targets sodium or chloride reabsorption of tubular cells, effectively preventing the establishment of the normal ion gradient by renal tubular cells.1‑3 Diuretics that increase net urinary excretion of sodium chloride or sodium bicarbonate also are referred to as natriuretic. The efficacy and use of each class of diuretics varies with their site of action and the mechanism by which sodium reabsorption is inhibited. The mechanisms of sodium resorption that are targeted by diuretic therapy vary in location within the renal tubule and include electrogenic passive diffusion (proximal and late distal tubule and collecting system); exchange with hydrogen (generated by the actions of carbonic anhydrase and bicarbonate reabsorption); co-transport with glucose, organic acids, and phosphate (proximal tubule); reabsorption along with chloride reabsorption (late proximal tubule); and co-transport with chloride and potassium (by way of the thick ascending limb of the loop of Henle, which results in formation of medullary interstitial hyperosmolarity).
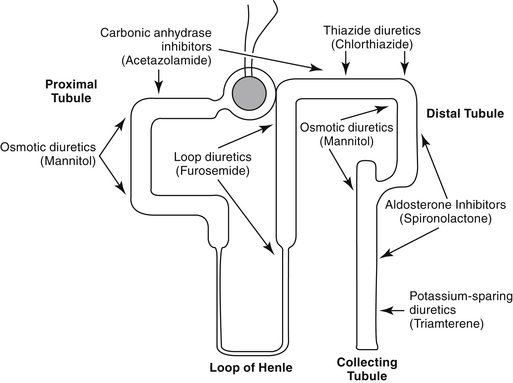
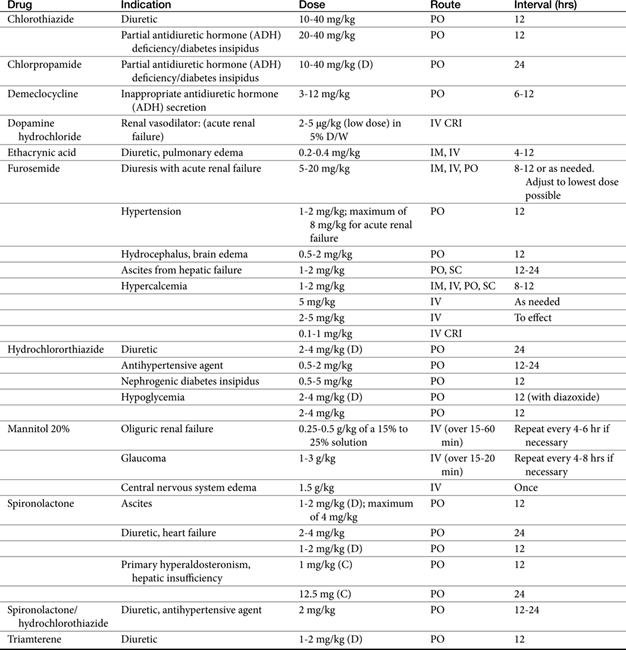
Principles of Diuretic Therapy Use
Several principles can guide diuretic therapy4: (1) The pattern of electrolyte excretion varies with the class of diuretic. (2) Maximal response to each drug class is limited by the site of action of the diuretic; as such, assuming drug delivery to the site of action has been appropriate, diuretic failure will occur to all members of the same drug class. class. (3) The combination of two (or more) diuretics with different mechanisms of action should cause additive and may cause synergistic effects.2
Factors Limiting Response to Diuretics
Several other factors may negatively affect response to diuretic therapy. Most diuretics are present at physiologic pH as uncharged molecules or organic ions and reach the renal tubular cell by active tubular secretion. The degree of ionization can affect the rapidity with which drugs are transported to renal receptors. Declining renal blood flow can preclude drug delivery to the site of diuretic action. For drugs that must reach distal sites (e.g., thiazides), it may be necessary to double doses to achieve a clinical response. For each diuretic a threshold (in drug concentration) must be reached before diuresis will occur. Lack of response may reflect simply an underdose for that patient, and dose titration is indicated. With renal disease characterized by proteinuria, many diuretics will remain bound to plasma proteins present in the tubular lumen and thus will remain inactive. Administration of the diuretic with albumin appears to facilitate response to diuretics in human patients with edema associated with the nephrotic syndrome. Resistance to diuretic therapy also may reflect the presence of another drug that decreases response. For example, nonsteroidal antiinflammatory drugs (NSAIDs) can alter intrarenal prostaglandin regulatory mechanisms, and a number of drugs compete for active tubular secretion of the diuretic into the tubular lumen.
In addition to their direct tubular effects, all diuretics indirectly influence renal tubular function. Accommodation to the effects of a diuretic in a normal animal may result in the loss of any pharmacologic effect several days after the start of diuretic therapy. Response to any diuretic will be modulated by internal homeostatic mechanisms that normally direct body fluid volumes and osmolar concentrations. For example, if a diuretic fails to cause a net sodium excretion (such as occurs with mannitol), ECF contraction will increase the concentration of electrolytes, stimulating water intake and replenishment of the lost volume.2 Refractoriness to thiazides can develop rapidly as a result of salt-retaining mechanisms being activated. Edema (ascites) associated with liver disease may not respond to diuretic therapy because signals from the cirrhotic liver indicate a depleted rather than an exaggerated ECF.2
Diuretics
Osmotic Diuretics
Osmotic diuretics increase the osmolality of extracellular fluid, enhancing flow of water from tissues to interstitial fluid and plasma. For a solute to act as an osmotic diuretic, it must be freely filtered at the glomerulus, not reabsorbed by the renal tubule, and be pharmacologically and metabolically inert. Mannitol (Figure 17-4) is the most commonly used osmotic diuretic; others include urea, glycerol, and isosorbide. As the concentration of an osmotic diuretic increases in the renal tubular lumen, osmotic forces overcome the movement of water with sodium into the renal tubular cell. Eventually, as water retention in the urine increases, sodium concentration decreases and passive sodium reabsorption is reduced. Sodium loss is, however, relatively small. Although mannitol appears to work throughout the renal tubule, the principal site of action appears to be the loop of Henle (see Figure 17-3).1 Mannitol is distributed to ECF and thus extracts water from intracellular compartments, increasing ECF, decreasing blood viscosity, and inhibiting renin release.1 The impact on intracellular compartments is therapeutically beneficial in patients with cerebral edema and glaucoma.1 Renal blood flow increases, removing NaCl and urea from the renal medulla and decreasing renal medullary tonicity. Medullary tonicity also may be reduced further by a prostaglandin-mediated increase in renal medullary blood flow. Mannitol is not absorbed after oral administration. In humans it is characterized by an elimination half-life of approximately 1 hour.
The major adverse effect of osmotic diuretics is increased ECF, which can be detrimental in patients with pulmonary edema or cardiac failure. Hyponatremia resulting from water extraction causes headaches, nausea, and vomiting in human patients.1 In contrast, loss of water in excess of electrolytes can cause hypernatremia and dehydration. Osmotic diuretics generally are contraindicated in anuria of severe renal disease or in patients that are not responsive to test doses. Mannitol and urea also are contraindicated for patients with active cranial bleeding. Glycerin (but not mannitol) can be metabolized, causing hyperglycemia.
Mannitol is most commonly used to treat acute renal failure resulting from an acute reduction in glomerular filtration or acute changes in renal tubular permeability. Mannitol provides protection to the tubules in that it attenuates reduction in glomerular filtration rate (GFR) associated with acute tubular nephrosis if the drug is administered before an ischemic insult.1 Efficacy (experimentally) for treatment of nephrotoxicity, however, is documented only when administered before the toxin; clinical efficacy is even less obvious. Mannitol is particularly indicated for treatment of toxic nephrosis because the concentration of the toxin in the urine will be reduced by the osmotic draw of water by the solute. In contrast to diuretics that act on tubular segments, osmotic diuretics usually maintain their effect in the oliguric state that accompanies acute renal failure because they will continue to be filtered by the glomerulus. If the tubular cell becomes permeable, however, as may occur with certain toxins or prolonged tubular ischemia, the osmotic diuretics may lose their efficacy. Yet in patients with acute tubular nephrosis, mannitol may convert an oliguric patient to a nonoliguric state.1
Mannitol is distributed to ECF and thus is not effective in movement of fluids from interstitial tissues. ECF volume will initially increase and may prove detrimental to the patient with decompensated cardiac function. Plasma osmolality increases after treatment with mannitol. Cerebrospinal fluid (CSF) and aqueous humor formation subsequently decrease. Whether mannitol will cross the blood–brain barrier is unclear. However, because mannitol is distributed into interstitial fluids but does not penetrate cell membranes, intracellular edema will also be reduced. As such, mannitol is used to treat selected causes of cerebral edema associated with increased intracellular fluid volume. The use of mannitol for treatment of acute brain injury is discussed in Chapter 27. A Cochrane review failed to find conclusive evidence regarding its efficacy, although the number of eligible clinical trials for review was limited.5 Mannitol can be used to decrease brain mass before neurosurgery. The use of mannitol to facilitate diagnosis of ureteral obstruction has been described.6
Carbonic Anhydrase Inhibitors
Two types of carbonic anhydrase are located in the proximal tubule, both targeted by carbonic anhydrase inhibitors: type II, located in the cytoplasm; and type IV, located in the luminal and basolateral membranes (see Figure 17-1). In the lumen, H+ (generated from the Na+-H+ transporter) reacts with HCO3− to form H2CO3, which, in the presence of brush border carbonic anhydrase, rapidly decomposes to water and CO2. The CO2 rapidly diffuses into the tubular cell, where it reacts with water to form H2CO3. This reaction normally proceeds slowly but is markedly accelerated by carbonic anhydrase in the cytoplasm. Because intracellular H+ is low (because of Na+-H+ co-transport), H2CO3 spontaneously ionizes to form H+ and HCO3−. An electrochemical gradient moves Na+HCO3– into the interstitial space, with water following. Chloride becomes concentrated in the lumen and diffuses down its gradient into the interstitium (see Figure 17-1).1 Carbonic anhydrase inhibitors target both the cytoplasmic and membrane-bound carbonic anhydrase, completely impairing NaHCO3 reabsorption in the proximal tubule (see Figure 17-3). The collecting tubule is a secondary target. As a result, urine concentrations of bicarbonate increase, and up to 35% of the filtered load is excreted. Because hydrogen ions are not generated by the conversion of bicarbonate to CO2 and H2O, they are not available for exchange with sodium. Thus the amount of acid and ammonia excreted in urine also decreases. The loss of titratable acid and ammonia secretion in the collecting duct results in an increase in urinary pH to approximately 8 and the potential development of metabolic acidosis.1 At least 65% of sodium bicarbonate is reabsorbed through carbonic anhydrase–independent mechanisms. Sodium and chloride not reabsorbed in the proximal tubule are delivered to the loop of Henle, where most of the chloride and sodium subsequently are absorbed. Up to 70% of potassium is excreted because of the increased sodium load. Bicarbonate remains in the urine, contributing to alkalinity. Ultimately, however, much of the bicarbonate that remains in the proximal tubular lumen is resorbed distally in the nephron by mechanisms that are not well understood.
Several sequelae result from the diuretic mechanism of carbonic anhydrase inhibitors. First, metabolic acidosis develops as bicarbonate is lost in the proximal tubules. Second, the filtered load of HCO3− decreases to the point that the uncatalyzed (spontaneous) reaction between CO2 and water leads to HCO3 reabsorption. This in turn decreases the response of the renal tubule to carbonic anhydrase inhibitors. Thus the diuretic effect of these drugs is self-limiting. However, in patients refractory to alternative diuretic therapy, the combination of acetazolamide with diuretics that block sodium resorption distally may result in marked naturiesis. Thus carbonic anhydrase inhibitors may be useful for combination therapy.1 In the distal tubule, as sodium is reabsorbed, potassium excretion markedly increases. Carbonic anhydrase is located in other tissues. Aqueous humor and CSF formation are both decreased by carbonic anhydrase inhibitors. Accordingly, the major indication for their use is glaucoma. The effect in the eye is direct and is not influenced by metabolic acidosis. The effect of carbonic anhydrase inhibition in the brain is not as well understood and may result from both the direct effects and the development of metabolic acidosis. Although carbonic anhydrase also is located in the gastric mucosa, only large doses of inhibitors reduce gastric acid secretion. Carbonic anhydrase activity in red blood cells will be impaired, causing an increase in CO2 levels in peripheral tissues and decreased levels in expired air. Carbonic anhydrase can increase delivery of solutes to the macula densa. Tubuloglomerular feedback may be triggered, increasing afferent arteriolar resistance and reducing renal blood flow and GFR.
Side effects of carbonic anhydrase inhibitors are not common. Large doses may cause drowsiness. Side effects can also result from urinary alkalinization or metabolic acidosis. Hepatic encephalopathy can be induced as renal ammonia is diverted from the urine. Precipitation of calcium phosphate may cause calculus formation. Respiratory or metabolic acidosis can be worsened. Carbonic anhydrase inhibitors are contraindicated in patients with cirrhosis or other causes of hepatic encephalopathy or conditions associated with acidosis. The impact of carbonic anhydrase inhibitors on urinary pH can reduce the rate of excretion of weak bases.1
Acetazolamide (Figure 17-4) is a potent, reversible inhibitor of carbonic anhydrase. Its primary indication in veterinary medicine is for the treatment of glaucoma with the intent to decrease aqueous humor formation. It is used less commonly to control CSF formation in patients with hydrocephalus or other causes of increased cerebral fluid pressure. Acetazolamide is characterized by some antiepileptic activity, although tolerance rapidly develops to this effect. Occasionally, acetazolamide might be used to alkalinize the urine. Indications might be for selected causes of crystalluria and to facilitate excretion of weakly acidic drugs, such as phenobarbital, and salicylate. Because its efficacy is self-limiting, combination with sodium bicarbonate may be indicated if persistent urinary alkalinization is desired.
Methazolamide is among the orally administered carbonic anydrase inhibitors used to treat glaucoma in dogs. Its impact on intraocular pressure and aqueous humor flow rate was described in Beagles receiving a single dose of 25 or 50 mg (10 dogs per group; 5 control) followed by multiple twice-daily dosing for 9 days.7 A diurnal difference in intraocular pressure was measured in all animals, being highest in the morning. At 25 and 50 mg, intraocular pressure decreased in the morning but increased in the evening compared with baseline. Compared with baseline, aqueous humor flow rate increased in both groups. Although response to 50 mg (compared with 25) was greater for both intraocular pressure and aqueous humor flow rate, the difference was not significant, perhaps reflecting a small sample size.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


