Chapter 4 Drug-Induced Diseases
Definitions and Predisposing Factors
An adverse drug event (ADE) is any harm caused by a therapeutic or preventive (or diagnostic) intervention (Figure 4-1). The evolution of the definition in human medicine during the past decade has led to further subdivision into medication errors and adverse drug reactions (ADRs).1 Medication errors are ADEs that result from a mistake made by the caregiver, including but not necessarily limited to administration of the wrong drug, dose, interval, or route to the wrong patient. Thus, ADEs are iatrogenic in origin. A potential subcategory of medication errors might be failure to modify a dosing regimen for recognized patient, drug, or disease factors that lead to inappropriately low or high drug concentrations at the site of action.
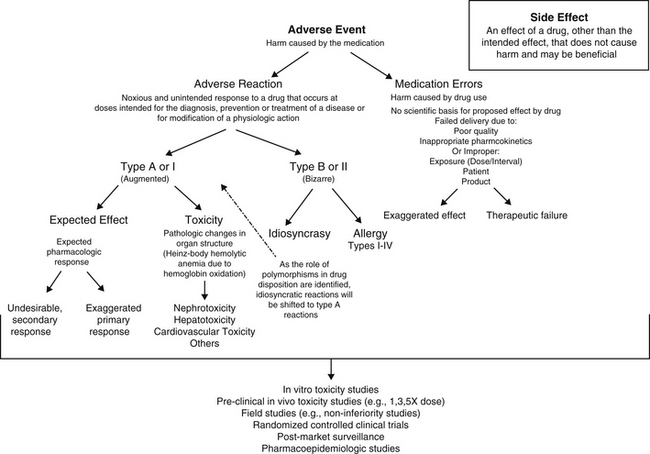
In contrast to medication errors, an ADR is a noxious and unintended response to a drug or other medication that occurs at a dose given with the goal of achieving the intended effect of the medication. As such, the term ADR implies a reaction that might cause serious harm to the patient and reflects a patient response to the inherent properties of the drug. An ADR may reflect a pharmacodynamic response or a pharmacokinetic effect. An ADR should be contrasted with the term side effect, which refers to an effect other than the intended effect that does not cause harm. A side effect may not be undesirable and may, in fact, be desirable or inconsequential to the health of the patient. For the purposes of this discussion, the term ADE will be used to refer to ADR with or without an iatrogenic basis. Adverse drug reactions can be further classified as either type A (type I) or type B (type II).2–4 Type A (“augmented”) adverse events generally result from drug concentrations at the site (generally estimated by plasma drug concentrations [PDCs] that either exceed the maximum or drop below the minimum therapeutic range (see Chapter 1). If the clinician is familiar with the drug and the patient, type A reactions are largely predictable and, as such, avoidable. Like type A, side effects are also generally predictable and dose dependent and for the purposes of discussion, will be included with type A adverse events in this chapter.
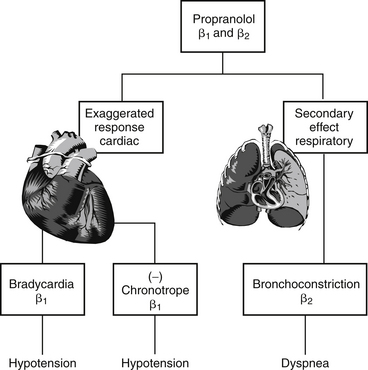
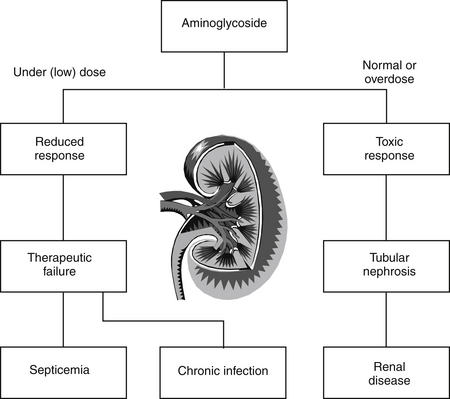
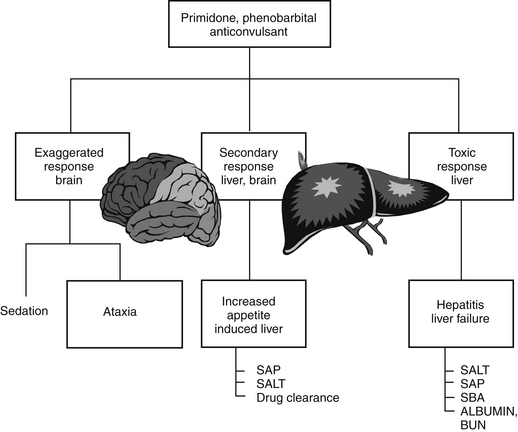
Generally, type A reactions are manifested as an exaggerated but normal or expected pharmacologic response to the drug (see Figure 4-1).5 This response may be the desired response (e.g., bradycardia in a patient receiving propranolol to slow a sinus tachycardia) but also may reflect an unwanted, secondary response resulting from the drug’s pharmacologic effects (e.g., bronchospasms induced by the beta-blockade effects of propranolol) (Figure 4-2). Some drugs also cause adverse events unrelated to their pharmacologic response. These reactions usually reflect damage to target cells and are referred to in this chapter as cytotoxic adverse reactions. Cytotoxic adverse reactions are exemplified by nephrotoxicity induced by aminoglycosides (Figure 4-3) or hepatic necrosis or methemoglobinemia induced by acetaminophen. Often, it is the metabolite of the drug rather than the drug itself that causes cytotoxicity (see Chapter 2). In such cases, drugs that induce metabolism, particularly in the liver (e.g., phenobarbital, phenytoin; Figure 4-4), may increase the risk of toxicity, whereas drugs that decrease metabolism may reduce the risk of toxicity (e.g., cimetidine).6–8 Cytotoxic drug reactions might be treated with drugs that scavenge radical metabolites (i.e., N-acetylcysteine, a glutathione precursor).
In contrast to type A events, type B (“bizarre”) events are not dose or concentration dependent. As a result, these reactions are not predictable and are largely unavoidable. They occur only in a small percentage of the population receiving the drug; in human medicine they account for approximately 6% to 12% of all ADRs.9 Generally, their incidence—indeed their existence—often is not documented until the drug is in wide use. In addition, because their cause is not well understood, treatment is generally limited to symptomatic therapy. Examples of type B adverse reactions include drug allergies or idiosyncrasies. Many of the idiosyncrasies eventually may be shown to be genetically or otherwise based (e.g., polymorphisms in transport or drug-metabolizing proteins [ivermectin toxicity of Collie-related breeds]), but the cause has yet to be identified and thus the reaction cannot be predicted. As with type A events, type B events may occur in response to the parent drug or its metabolite. This chapter will focus on allergies as type B ADRs.
The impact of ADEs on the monetary and health costs in human medicine were assessed in the late 1990s. The implementation of a voluntary, anonymous reporting system has facilitated medication error reporting in human medicine and thus enabled a more accurate assessment of their impact.10 The majority (66%) of medical errors occurred during transfer of patient care within hospitals, and another 19% occurred on discharge. In a human intensive care unit (ICU) environment, 1 out of every 5 doses of medication was associated with an ADE as a result of medication errors. The veterinary profession has yet to implement a mechanism whereby ADEs due to medication errors can be assessed. Yet needs are equally applicable to veterinary medicine.10
Organs most susceptible to type A drug events usually are those subjected to the greatest exposure or concentration of the drug. Thus the organs with the greatest blood flow and those organs capable of drug concentration, such as the liver and kidney, are the most vulnerable to systemic drugs. Highly metabolically active organs are also more likely to manifest toxic effects for two reasons. First, such organs depend on the presence of energy, and anything that impairs acquisition of energy (including blood flow) can lead to malfunction. Second, if the metabolic activity includes metabolism of compounds, the production of potentially reactive metabolites can increase the likelihood of cytotoxicity if these metabolites interact with cellular structures. In contrast to Type A ADEs, the organs most susceptible to damage by type B reactions tend to be the organs that contain tissues that act as haptens for drug-induced allergy (e.g., skin, blood-forming units) or tissues that filter and trap immune complexes (e.g., glomerulus, joints). Organs containing a preponderance of mast cells also are more likely to manifest immune-mediated reactions (i.e., “shock organs”; see the section on allergic reactions). A summary of compounds causing predominantly type A drug reactions and, when available, their antidotes, can be found in Appendix 5. A variety of factors can influence the likelihood of adverse, and particularly toxic, reactions.6–8 Factors that predispose a patient to the development of type A adverse events are discussed in Chapter 2.
Not all adverse reactions are clinically evident. Sometimes the reaction is not detectable unless actively sought. For example, clinical laboratory tests may detect a drug-induced hepatotoxicity (e.g., increased serum alanine transferase activity) that is otherwise clinically silent. Many drugs can directly interfere with or indirectly influence clinical laboratory tests, including endocrine function testing (discussed later).11 Organ predisposition to drug-induced toxicity may show diurnal variations. For example, both aminoglycosides and cisplatin12 exhibit increased renal toxicity in humans when administered in the evening as opposed to in the morning. For the aminoglycosides, safety in the morning has been attributed, in part, to the increase in glomerular filtration rate that occurs in the morning;13 an increased sensitivity to interleukin-6–induced inflammation has been suggested for cisplatin.
Principles of Toxicology Relevant to Pharmacology
Terminology of Toxicity and Safety Assessment
Toxicology refers to the study of poisons and their effect on living organisms.14 A poison is any substance that is injurious to animals; the term is synonymous with toxic substance, toxic chemical, and toxicant. It is important to note that any drug can become a poison (“the dose makes the poison”), and the toxic response to the poison tends to correlate with the dose (or duration). Toxic response refers to the effects manifested by an organism in response to a toxic substance. Toxicity describes the quantitative amount or dose of a poison that produces a toxic response or effect. Acute toxicity generally results from a single dose or exposure or multiple doses in a 24-hour period. Most acute toxicants rapidly interfere with critical cellular processes. Subacute and subchronic toxicity occurs after 1 week to 1 month of exposure. Chronic toxicity occurs after 3 or more months of exposure.14 These latter terms are generally applied to humans, but the relative duration of exposure applies equally to animals. Toxic chemicals can act directly by injuring the cells with which they come in contact or indirectly by injuring a group of cells that subsequently precipitate injury to others. Alternatively, toxins can act indirectly by interfering with a physiologic process on which a group of cells are vitally dependent. Toxins can act systemically (the majority), locally, or a combination of the two.15
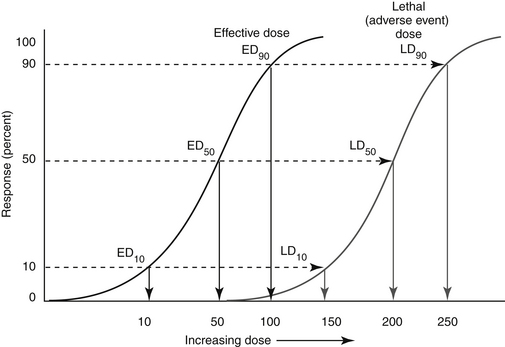
An indicator of the assessment of toxicity is the LD50, or the dose per unit weight of a chemical that kills 50% of animals receiving it (the median lethal dose) (Figure 4-5). The LD50 is both drug and species specific. The lethal dose of a drug can be compared with the dose necessary to induce the desired pharmacodynamic response (effective dose, or ED) in a targeted percent of the sample population (see Chapter 1). The ED50 is the dose expected to induce the response in 50% of the population; its more useful companion indicator is the EC50, which describes the concentration at which 50% of individuals respond, thus removing the effects of drug absorption and distribution from consideration. Like the LD50, ED50 and EC50 are generally based on single dosing and do not take into account duration of exposure. The therapeutic index offers a measure of the relative safety of a drug by comparing the dose producing toxicity to that causing the desired effect, or the ratio of the LD50 to ED50 (see Figure 4-5). A related term is margin of safety which is the ratio minimal lethal dose (LD01) and the dose associated with the greatest efficacy (ED99). Caution is recommended if the margin of safety (MOS) is less than 1. Alternatively, rather than measure lethality as the adverse event, the concentration necessary to cause a specific adverse event can be measured and used to calculate the therapeutic index. A second set of terms by which the safety of a compound might be assessed are the lowest observed adverse effect level (LOAEL) and no observed (adverse) effect level (NOEL, or NOAEL). The LOAEL is the lowest concentration of drug associated with observed adverse effects. The NOAEL is defined as the greatest concentration of a compound that causes no detectable adverse effect. When compared with anticipated drug intake, it provides an alternative margin of safety. The absence of these effects can be used to help verify the NOAEL. The NOAEL generally is based on chronic exposure and thus may be more relevant for drugs administered at more than a single dose. NOELs and LOELs (no or lowest observed effect level, respectively) differ in that they may not indicate an adverse effect, but in fact may address a beneficial effect. All four terms are important to risk assessment which addresses exposure and the adverse outcomes that might be associated with exposure (see later discussion).
Other toxicologic terms relevant to drugs include teratogen, which is a compound that causes abnormal fetal development. It is important to note that teratogenicity is not the only form of toxicity that may occur in utero; any toxic effect that occurs in the adult is likely to occur in the developing fetus as well. Carcinogenesis refers to the ability of a compound, or a carcinogen, to cause cancer. Cancer cells are cells that have been able to avoid the sophisticated mechanisms that control normal growth, development, and division. Induction of cancer by a compound involves many variables, including duration, dose, and frequency of exposure. Generally, carcinogens take 20 years or more to induce cancer, and the cause-and-effect relationship between the compound and the cancer often is not recognized.15,16 Although some compounds can directly interact with DNA, leading to a cancerous cell, most compounds must first be converted to a reactive metabolite to covalently bond with DNA. Damage may still be avoided if DNA repair occurs before cell division. Cellular damage increases the stimulus for division of adjacent cells, leading to a new cell type with new genotypic and phenotypic properties that can then be transformed to a malignant cell under the correct conditions. A number of compounds are recognized to be initiating agents, targeting molecular DNA, whereas others are considered promoters, acting to increase the incidence of cancer or decrease the latency period without interacting with DNA. These latter compounds must be administered repeatedly and after the initial insult. Endogenous compounds such as growth factors or hormones may act as promoters.15
Two mechanisms by which drugs can cause cell death are apoptosis and necrosis, each with distinct morphologic and biochemical characteristics. Apoptosis is an active process characterized by cell shrinkage, nuclear and cytoplasmic condensation, chromatin fragmentation, and phagocytosis. In contrast, necrosis is a passive process resulting in inflammation associated with cellular and organelle swelling, rupture of the plasma membrane, and spilling of cellular contents into the extracellular milieu.17 Because apoptosis is an active process, sufficient intracellular energy must be maintained; depletion of adenosine triphosphate (ATP) may cause an apoptopic process to become a necrotic process. Not surprisingly, mitochondria appear to play a role in apoptosis. A number of toxicants cause their effects by disrupting mitochondria function and thus ATP production. However, several drugs appear to exert their toxicologic effect through induction of apoptosis (e.g., digoxin, selected chemotherapeutic agents).17
Genetic diversity is increasingly being identified as a cause for individual variation in response to toxins and drugs, leading to the fields of toxicogenetics and pharmacogenetics, respectively.5,16,18 The role of polymorphic cytochrome P450s as a cause of interindividual differences in xenobiotic metabolism and drug toxicity is fairly well established. In humans polymorphism occurs less commonly in those cytochrome P450s responsible for carcinogen activation (CYP1A1 and 2, CYP1B1, CYP2E1, and CYP3A4) compared with those that are primarily responsible for drug metabolism (CYP2C9, CYP2C19, CYP2D6, and CYP3A4). Polymorphisms in drug-metabolizing enzymes have been associated with drug hypersensitivities.6 All hepatic drug-metabolizing P450s are polymorphic with the clinically most important polymorphism in humans occurring with CYP2C9, CYP2C19, and CYP2D6.19 Polymorphisms in cytochrome P450s of animals is now being elucidated but may play a role in adversities in certain breeds (e.g., Beagles, Greyhounds). In addition, polymorphism in P-glycoprotein (P-gp) is a well-established reason for susceptibility to adverse drug events for selected drugs in Collie-related breeds (see Chapter 2).
Postmarket Surveillance
The approval process for human-marketed drugs is designed to identify adverse reactions at several stages preclinical phase (short- and long-term animal testing): through phase III (extended clinical trials) and continuing into phase IV, which includes a mandated postmarket surveillance. However, the drug approval process for animals is not as regimented (see Figure 4-1). The requirements by the Food and Drug Administration’s Center for Veterinary Medicine tend to vary with the drug undergoing approval (See Appendix 1).20 In general, directed toxicity studies that are implemented during the approval process involve a small number (e.g., 2 to 30 but most commonly 4 of each sex) of study animals receiving the drug under conditions of exaggerated use—that is, doses (e.g.,1, 3, 5× the highest labeled dose,) and durations (e.g., 3×) of several magnitudes greater than the anticipated approved intended use. Field trials implemented during the approval process are performed in the approved species, generally as controlled (placebo or positive), randomized clinical trials, under conditions of intended use. Such studies generally involve a much larger sample population (e.g., hundreds of animals) than the directed toxicity studies. However, although toxicity studies do predict a number of adverse events that occur in the target species,21 neither directed toxicity nor field studies are likely to involve a sample size sufficiently large to allow detection of many adverse events that occur in a very small proportion of patients (e.g., 1 in 1000 or more). Detection of such an adverse event is likely to require thousands of subjects. Further, directed studies are generally performed in a sample population of generally healthy animals unaffected by the complexities of host, drug, and disease factors associated with drug use in the target population (see Chapter 2). As such, postmarket surveillance studies and pharmacoepidemiologic studies (which focus on subgroups of target species) are particularly important to the safety assessment of approved drugs used in animal patients.21 For the same reason, if an ADE is suspected as a result of drug therapy, the importance of reporting it (or suspicion of it) cannot be overemphasized. Note that safety assessment of human-marketed drugs are studied in dogs but with an emphasis on human rather than animal safety. Although all approved human-marketed drugs are likely to have been studied in dogs during the preclinical phase, most of this information is not generally available unless specifically requested through Freedom of Information Act mechanisms. If an ADE is suspected, it first should be reported to the manufacturer; by law, animal drug pharmaceutical companies must report adverse events reported in animals to the Food and Drug Administration (FDA). The veterinary profession, probably more so than the human-medicine community, is less likely to report adverse event perhaps because the mechanism by which the information is forwarded to the veterinary health care provider is limited in both scope and distribution. In contrast to drugs, adverse event reporting for animal dietary supplements is entirely voluntary, with no obvious mechanism for reports to be collected, assessed, or returned to the veterinary profession. It is only through postmarket surveillance that more clinically relevant assessments of drug safety can emerge, and subtle differences in the safety of different drugs might become apparent. Assessments include hazard or risk assessment, which express the probability of harm under conditions expected with use of the drug. Hazard is the potential to cause harm—that is, the inherent toxic nature of the compound. Risk is the likelihood that harm will occur. As such, it takes into account both hazard and exposure—that is, the amount of the toxin ingested and thus dose and duration.22 Risk–benefit analysis compares the risk associated with the use of a product and its potential benefits.23 As such, a risk/benefit ratio addresses the acceptability of an adverse reaction by taking into account the importance, frequency, and duration of therapeutic benefit and the adverse reaction. Risk–benefit analysis generally is based on postmarket use of a product and a mechanism of surveillance that detects and assesses adverse events.24
Among the difficulties in assessing risk through postmarket surveillance is the attribution of the adversity to the drug. Information submitted in an ADE report is limited, and care must be taken to not assume a cause–effect relationship The FDA’s Center for Veterinary Medicine has recently reviewed its ADE reporting program.25 It defines an ADE as an undesired or lack of desired response to a drug, medical device, or (in food animals) medicated feed. As such, a distinction is not made between adverse events associated with medication errors and those associated with inherent properties of the drug. In its 2004 report, the FDA indicates that a 6-point scoring system evaluates drug reactions submitted from manufacturers, pet owners, or veterinarians. Information collected includes previous experience with the drug (i.e., historical evidence of adversities from the label information or previously submitted reports), timing of the event in relation to dosing, alternative causes, the role of overdose, and effects of dechallenge or rechallenge. Plans for improved submission include a web-based submission process.
The FDA website (http://www.fda.gov/AnimalVeterinary/SafetyHealth/ProductSafetyInformation/ucm055375.htm; accessed May 2010) can be reviewed for yearly and cumulative adverse event reports that have been reported in animals. Unfortunately, at the time of publication, the reports provide no evidence of frequency of occurrence, other than a ranking. This decision was made in part because of the inability of the FDA to standardize the number of adverse events by the number of doses administered (or units sold).
Predicting Drug Safety
Woodward21 has demonstrated that, to some degree, studies implemented during the approval process tend to predict adversities that emerge in target species, but with marked limitations. Guengerich26 has discussed the role of predictive toxicology based on drug chemistry in assessing clinical safety of drugs. Toxicities are likely to be predictive if the drug is characterized by a high level of intrinsic toxicity; for such drugs chemistry may often predict toxicity. Toxicity is less predictable, although still reasonably so if metabolism plays a role, but is much less predictable if the toxicity is idiosyncratic (i.e., type B or II).
Idiosyncratic Reactions: Allergic Drug Events
The clinical manifestations of idiosyncratic ADRs vary with the type of reaction and the body system targeted. Generally, for allergies, previous exposure to the drug must occur regardless of the type of reaction, or therapy must have been sufficiently long (i.e., at 10 to 14 days for some drugs) for an allergic response to develop. However, exceptions appear to exist, as is exemplified by allergy-based reactions to sulfonamides, which may occur in as early as 5 days (see Chapter 7). Drugs generally are too small in molecular size (<1000 D) to be sufficiently antigenic. As such, drugs that induce an allergic response generally act as haptens, covalently combining with a body tissue that then also becomes antigenic. As a result, the allergic response may be directed toward the drug or tissue.9 The hapten hypothesis is controversial because only a small percentage of persons develop a reaction, possibly because of a failure to develop tolerance. Failed tolerance may occur at one or more proposed sites. First, the role of metabolism in the formation of chemically reactive metabolites as the initial step in mediating idiosyncratic drug responses (including allergies) is increasingly supported by scientific studies. Those individuals that produce more metabolites appear to be more likely to fail to develop immune tolerance to a drug.9 For the same reason, the dose of antigen exposure may determine the type of response. The role of CYP enzymes in idiosyncratic reactions (whether or not allergy based) is increasingly being recognized. Second, the metabolite must bind with an appropriate ligand, one with a high epitope density sufficient to induce an immune response. Third, an allergic response to a drug requires activity of an antigen-processing cell. As such, it may be more likely in the presence of inflammation, such as might be the case with a viral or bacterial infection or after stress or traumatic damage. Molecular signals that activate immune cells may also be more prevalent in the face of large concentrations of reactive metabolites because of their ability to induce oxidative stress.9 Fourth, other factors that may determine emergence of an allergic response (i.e., failed tolerance) may include failed downregulation of regulatory factors. A balance toward protective t-helper (Th1 or 2) response may preclude emergence of an allergic response; a loss of balance may facilitate it. Accordingly, both genetic predisposition and environmental factors contribute to emergent drug allergies.9
Type I allergic reactions (immediate or anaphylactic) are IgE mediated and result from the release of chemical mediators (e.g., histamine, serotonin, eicosanoids) from tissue mast cells or basophils. The reaction occurs within minutes after drug administration regardless of the dose administered (Figure 4-6). Clinical manifestations generally include nausea, vomiting, circulatory collapse, tachycardia, pulmonary edema, and neurologic signs. Urticaria and angioedema may also be evident. Clinical signs may be species dependent, depending on the shock organ of the species. The shock organ generally is the organ in which mast cells occur in greater numbers. In the dog the shock organs tend to be the liver and gastrointestinal tract; in the cat the shock organ generally is the lung, with fulminating pulmonary edema being a clinical manifestation.
The exact antigen that causes anaphylaxis may not be known. For example, even though anaphylaxis in microfilaremic dogs after administration of microfilaricides is well documented, the specific antigen released by the effect of the drug is not known.27 When given to microfilaremic dogs, both dimethylcarbamazine and ivermectin can induce shock manifested as peripheral vascular collapse, dyspnea, bloody diarrhea, and other clinical signs and laboratory test results consistent with anaphylaxis.
Treatment of drug-induced anaphylaxis is directed toward prevention of the physiologic response to mediator release (i.e., epinephrine and antihistamines) and prevention of further histamine release (e.g., epinephrine and glucocorticoids; possibly antihistamines). Ideally, antihistamines might also include the newer classes that may decrease mast cell degranulation as well as block H1 receptors associated with histamine-mediated shock. Interestingly, and perhaps disconcertingly, glucocorticoids themselves have been associated with anaphylactic reactions (see Chapter 30).28 Supportive therapy is also indicated. Prophylactic pretreatment in cases of anticipated anaphylaxis helps decrease the manifestations of anaphylaxis by decreasing the mast cell response. Drugs associated with type I allergic reaction in humans include penicillins, angiotensin-converting enzyme inhibitors (particularly in the first 3 weeks of therapy), nonsteroidal antiinflammatories drugs (NSAIDs), and opioids. However, the latter drugs may actually be more associated with an anaphylactic-like reaction, also referred to as an anaphylactoid reaction.
An anaphylactoid reaction is very similar to anaphylaxis but differs in that it is not mediated by an antigen-IgE response and thus is not allergic or immune mediated. Rather, selected drugs or compounds cause direct mast cell degranulation. Generally, these drugs are cationic (basic) and include opioids (particularly morphine [see Figure 4-6], polymyxin, radiographic contrast agents, thiacetarsamide, and amphotericin B). Hyperosmolar solutions such as mannitol can also cause direct mast cell degranulation. Adverse response to rapid intravenous administration of enrofloxacin, which is both hyperosmolar and basic, may represent an anaphlylactoid response or may simply reflect (in cats) stimulation of the chemoreceptor trigger zone. However, ciprofloxacin has been associated with an allergic response in humans,29 and the author is aware of at least one dog treated with ciprofloxacin for which the adverse reaction met the World Health Organization category of “probable” ADR. Anaphylactoid reactions tend to be related to dose. As such, administration of a small test dose may help the clinician determine the likelihood of occurrence. Response is probably more likely with intravenous administration. In addition to the prophylactic measures for anaphylaxis, decreasing the rate of drug administration may help reduce the risk of the adverse response.
Type III drug reactions (immune complex disease, or serum sickness) are induced by antigen–antibody complexes involving either IgG or IgM and complement activation. Circulating antigen–antibody complexes may be filtered by and lodged in the vasculature of a number of organs, including the kidney, central nervous system (CNS), or peripheral vasculature. Clinical signs generally refer to the predominant organ affected but also include fever and lymphadenopathy. The Arthus reaction is a variation of the type III reaction and is manifested as swelling and pain at the site of drug administration. Among drug reactions in veterinary medicine, the potentiated sulfonamides are probably the most well-recognized cause of type III immune-mediated drug reaction.30 Type IV drug reactions (delayed hypersensitivity, cell mediated) reflect cellular response at the site of the antigen. Lymphocytes and macrophages infiltrate the site and cause mediator release that perpetuates the inflammatory response.
Among the drugs recognized to be associated with drug allergies in dogs are the sulfonamides. Many medications contain a sulfonamide (a sulfur dioxide [SO2] and nitrogen [N] moiety), including sulfonamide antimicrobials (derivatives of sulfanilamide in which the sulfonamide is attached to an aryl amine; e.g., sulfamethoxazole, sulfadiazine, sulfadimethoxine), “coxib” cyclooxygenase-1–sparing NSAIDs (e.g., deracoxib, firacoxib), carbonic anhydrase inhibitors (e.g., acetazolamide), diuretics (e.g., hydrochlorothiazide, chlorthalidone, furosemide), uricosurics (e.g., probenecid), drugs to treat inflammatory bowel disease (e.g., sulfasalazine), sulfonylureas (e.g., glyburide, glipizide), and selected anticonvulsants (e.g., zonisamide).31 However, it is likely that a metabolite associated with the nitrogen moiety is responsible for the reaction, with reactions being limited to molecules containing an aryl-amine (both the sulfur and an amine moiety are attached directly to a benzene ring; sulfonylarylamine), a structure limited to sulfonamide antimicrobials. The underlying pathophysiology, clinical manifestations, and other aspects of the response have been well described32,33 and are reviewed in Chapter 7.
Adverse Drug Events by Body Systems
Drugs causing adverse events in the various body systems are listed in tabular form in the respective sections on body systems. Discussion of the adverse reaction also can be found in the appropriate chapter. When available, treatments are offered, including tabular presentation (see Appendix 5).
Liver
The liver is vulnerable to drug-induced toxicity for several reasons (Box 4-1).34–37 The potential for hepatotoxicity can be enhanced by dietary imbalance (high fat, low protein), presence of disease concurrent with administration of drugs that alter hepatic drug-metabolizing enzymes or hepatic blood flow,37 and age.38
Box 4-1 Hepatic Drug-Induced Toxicity
The liver is vulnerable to drug-induced toxicity for several reasons:
Drug-induced and chemical-induced liver injury have been classified into two categories.34,35,37 Type I toxins, or intrinsic hepatotoxins, cause type A adverse events, which are predictable, dose and time dependent and occur in most, if not all, subjects exposed to appropriate doses of the substance. Any drug metabolized by the liver probably can cause some degree of type I hepatic disease simply by the production of phase I metabolites, which as a general rule tend to be toxic because of their reactivity. The longer the drug is used and the higher the dose, the more likely the ADE will occur. Type II, or idiosyncratic hepatotoxins, cause type B events, which are unpredictable and dose and time independent (consistent with Type B). Their occurrence is sporadic and not reproducible.
Drug-induced hepatotoxicity (Box 4-2) is associated with a wide range of histologic changes, from acute, reversible, and clinically benign lesions to those that cause fatal massive necrosis, chronic hepatitis, or malignancy.34,35 Some drugs characteristically cause only a single lesion, whereas others cause multiple lesions. The lesions caused by any drug are rarely specific for that drug but can be caused by a variety of drugs or other disorders, often limiting the potential usefulness of biopsy (Figure 4-7).
Box 4-2 Examples of Drugs or Drug Classes Associated with Hepatotoxicity∗†
∗ Many other drugs that are metabolized by the liver are potentially hepatotoxic because of the production of phase I reactive metabolites.
† Microscopic lesions tend to be centrolobular in location, associated with necrosis, but otherwise nonspecific for most drugs.
‡ In the author’s experience, marked increase in serum alkaline phosphatase may occur but is not necessarily associated with hepatic dysfunction.
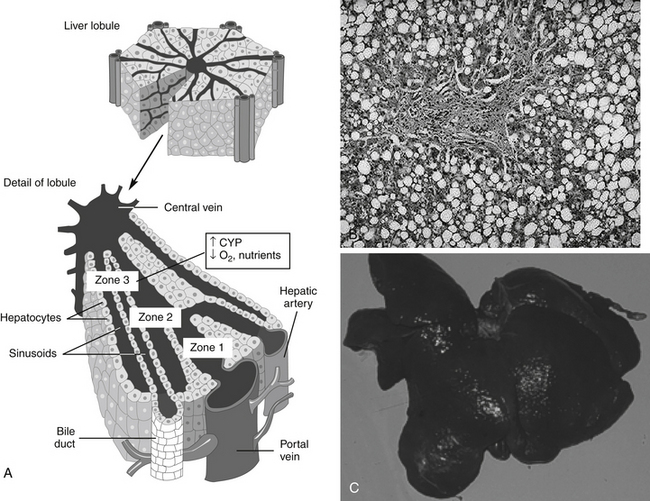
Frequently, drug-induced hepatic injury is limited to select regions or zones (e.g., central, middle, or peripheral) in the lobule.34,35 Various histologic lesions associated with drug hepatotoxicity have been described.34,35 Zonal necrosis usually results from type I or predictable toxins. The production of toxic metabolites may be an important cause of zonal necrosis because drug-metabolizing enzymes predominate in zones most likely to develop necrosis. In most cases of acute injury, the process is either fatal or completely resolved. If exposure is chronic or recurring, however, the lesions may persist and progress, depending on the dose, agent, and health of the patient. Lipid accumulation, usually of triglycerides, may be associated with either minimal alteration of hepatic function or with both clinical and laboratory manifestations of liver dysfunction.
Drug-induced cholestasis is not well understood. Drugs can target bile ducts or canaliculi, causing primarily cholestasis without hepatocellular disease. When accompanied by an inflammatory infiltrate, systemic illness usually occurs, whereas cholestasis without inflammation is associated with no or very mild clinical signs. Recovery usually occurs after discontinuation of drug therapy.34,35 Drugs can also affect primarily sinusoidal or endothelial cells, causing primarily fibrosis or veno-occlusive disease. Veno-occlusive disease tends to be predictable and is most commonly associated in people taking anticancer drugs. An immune basis has been recognized for some drugs causing clinical signs consistent with chronic active hepatitis.
Treatment of drug-induced liver disease is primarily supportive. Because reactive metabolites often either cause or exacerbate disease, however, use of compounds that help prevent metabolite damage to the liver should be considered. Specific examples include N-acetylcysteine, a precursor to intracellular glutathione; ascorbic acid, another type of oxygen radical scavenger; S-adenosylmethionine (SAMe), a compound that contributes to a number of methylation reactions in the body; and the herbal agent silymarin (see Chapter 19). Care must be taken to ensure that the duration of treatment exceeds the duration of activity of the toxicant, as has been demonstrated for N-acetylcysteine for treatment of acetaminophen toxicity.39
Hepatotoxic Drugs
Inhalant Anesthetics
Adverse events to inhalant anesthetics are unusual in veterinary medicine,40,41 in part because duration of anesthetic exposure is limited. Historically, methoxyflurane administration in dogs has occasionally been associated with acute centrilobular necrosis accompanied by a mixed inflammatory infiltrate. Halothane-associated hepatic injury in the dog has not been confirmed, although a clinical case report has described acute hepatic necrosis after its use. In humans the degree and incidence of halothane-induced liver damage do not appear to correlate with the duration or number of exposures and therefore has been suggested to reflect an idiosyncratic hypersensitivity.
Mebendazole and Oxibendazole
The bendazole anthelmintics have been associated with liver pathology. Acute centrilobular hepatic necrosis and fatal fulminating hepatitis have been reported in dogs after the clinical and experimental administration of the anthelmintics mebendazole and oxibendazole.42,43 Clinical signs were evident in as few as 2 days or as many as 10 to 14 days after administration. Although mebendazole was originally thought to be an intrinsic hepatotoxin, other studies suggest that it produces an idiosyncratic reaction.
Sulfonamides
Sulfonamide antimicrobials are associated with toxicity of multiple organs, including the liver.30,32,33,44 Sulfonamides do not appear to differ in their likelihood of causing toxicity. In one report that supports an idiosyncratic reaction, the duration of therapy before hepatotoxicity developed ranged from 4 to 30 days, and the dose ranged from 18 to 53 mg/kg every 12 hours.44 The mechanism of sulfonamide toxicity is discussed in more depth in Chapter 7.
Thiacetarsamide and Melarsomine
Thiacetarsamide (caparsolate) is associated with hepatic injury in humans and animals. Chronic exposures in humans are more likely to cause clinically significant hepatic disease. Hepatotoxicity is, however, a common complication of acute administration of thiacetarsamide for heartworm disease in dogs, although residual effects after therapy is completed are not expected (see Chapter 14). In normal animals melarsomine causes less hepatotoxicity and renal toxicity than thiacetarsamide.45
Xylitol
The sugar alcohol xylitol is associated with life-threatening hypoglycemia and hepatic necrosis in dogs. Most cases, reported by the American Society for Prevention of Cruelty to Animals (ASPCA) Poison Control Center, reflect over ingestion of products containing xylitol as a sweetener (a small to average cookie may contain approximately 4 g xylitol). In contrast to humans, xylitol induces a ten-fold increase in insulin secretion in dogs compared to an equivalent amount of glucose. The result is a precipitous drop in serum glucose within 30 to 60 minutes after ingestion of as little as 100 mg/kg. Onset of hypoglycemia may be offset for 12 hr if xylitol-containing gum is ingested. Lethargy, ataxia, collapse, and seizures may occur. Clinical pathology may also reveal hypokalemia as potassium moves into the cell with glucose, and hypophosphatemia as a result of insulin’s effects on cell permeability.45a,b The impact on cats is not clear. More recently, hepatic necrosis has been reported in dogs ingesting xylitol. Enzyme increases within 12 to 24 hours after ingestion of 500 mg/kg or more are followed by clinical signs and sequelae, including coagulopathies consistent with acute hepatopathy. Hyper- rather than hypophosphatemia associated with acute hepatopathy may be a poor prognostic indicator. Sorbitol does not appear to be associated with toxicity in dogs.
Kidney
Like the liver, the kidney is vulnerable to drug-induced toxicity for several reasons (Boxes 4-3 and 4-4).46 Specific cellular or subcellular sites of nephrotoxins frequently are not known. Usually, a toxin affects more than one type of renal tissue because of the high drug concentrations to which the kidney is exposed. The glomerulus is susceptible to direct nephrotoxicity as well as to indirect toxicity such as that caused by immunologic injury.46 Many nephrotoxins cause predominantly proximal tubular damage. This is expected in part because blood flow is greatest in the renal cortex, where the proximal tubules are located. Variations in proximal tubular susceptibility to toxins may reflect different tubular functions.46,47
Box 4-4 Renal Drug-Induced Toxicity
The kidney is vulnerable to drug-induced toxicity for several reasons:
A study in human medicine focused on the use of nephrotoxic drugs in the critical care patient.48 Reductions in renal blood flow associated with hemodynamic responses to a myriad of illnesses predisposes the ICU patient to acute renal failure which occurs in 6% of human ICU patients. Prolonged use of vasopressors increases the risk of renal hypoxia. Additionally, of note, the use of low-dose dopamine (≤3 μg/kg/min) as a nephroprotectant, particularly in patients with acute renal failure (representing at least 6% of ICU patients) was discouraged. Although renal vasodilation and urine flow increase, outcome does not improve, and the increased risk of cardiac or other adversities balances any potential nephroprotection.48 NSAIDs were cited as a particular risk in ICU patients; newer drugs that target COX-2 do not appear to offer an advantage in regard to nephrotoxicity. Nephrotoxicity induced by NSAIDs in the critical care environment occurs rapidly and is manifested as a rapid increase in serum creatinine. If an NSAID must be used in the ICU patient, one with a short half-life is recommended, and use of other nephroactive drugs (those that alter renal blood flow) are discouraged.48 The ICU patient also is at increased risk for aminoglycoside toxicity, with urine enzymes the earliest indicator. Clinical evidence of nephrotoxicity occurs within 5 to 10 days of therapy; once-daily (or less frequent) therapy reduces the risk. Likewise, amphotericin B often is associated with acute renal dysfunction. Sodium-containing fluids and lipid-based products are recommended in the patient at risk. The use of nacetylcysteine to protect the kidney should be considered for selected drugs.
Nephrotoxic Drugs
Most nephrotoxic drugs are discussed in relevant chapters. Methoxyflurane causes a dose-dependent, high-output nephrotoxicity in humans. Toxicity appears to be the result of oxalate metabolites and inorganic fluoride. Oxalate metabolites crystallize in and obstruct the tubules, whereas inorganic fluoride produces tubular necrosis.42 Veterinary reports of methoxyflurane-induced nephrotoxicity are rare, probably because veterinary patients are at a reduced risk of developing nephrotoxicity because exposure (surgery) times are much shorter than in humans.49
Trivalent arsenicals such as thiacetarsamide denature proteins by binding to sulfhydryl groups. The glomerulus is often the first site of arsenical-induced nephrotoxicity, but proximal tubule damage predominates, probably because of the large number of enzymes that are denatured in this region.46 Initial proteinuria is followed by tubular necrosis and degeneration.
Gastrointestinal
Stomatitis may progress to ulcerations with several drugs, particularly antineoplastic agents. Those most likely to cause stomatitis are listed in Table 4-1. A number of drugs are sufficiently caustic that ulcerations occur if the drug remains in contact with the mucosa (see Table 4-1). A number of drugs, including doxycycline and other drugs administered orally as a tablet have been associated with local mucosal damage and subsequent esophageal strictures in cats (see Chapter 19).
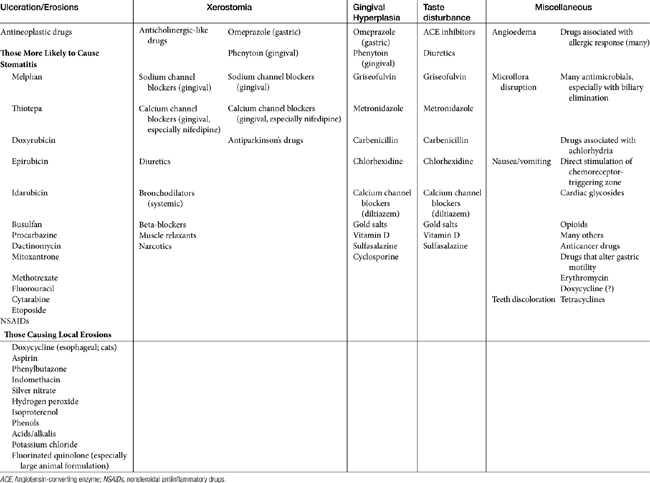
Xerostomia (dry mouth) has been associated with anticholinergics and drugs with anticholinergic-like effects (see Table 4-1), as well as other drugs. Taste change is more discernible in humans and is caused by a number of drugs, including several antimicrobials (see Table 4-1).
Gingival hyperplasia has been reported for several drugs, including phenytoin and (in humans) calcium or sodium channel blockers.50 The risk of its emergence might be reduced with good dental hygiene; in dogs it responded to metronidazole therapy.51 Gingival hyperplasia is a recognized side effect of cyclosporine administration in dogs and cats (package insert, Atopica); 31% of the dogs developed gingival hyperplasia and gingivitis, which responded to metronidazole and spiramycin.51 However, the more common effect of drugs on the oral mucosa is erosion. A number of drugs cause direct erosion with prolonged contact, especially in the feline esophagus (Table 4-1). Any drug that is antianabolic or inhibits cellular division is potentially toxic to the gastrointestinal tract by impairing the rapid turnover of epithelial cells in the mucosa (see Table 4-1). Tetracyclines and chloramphenicol are antianabolic, although long-term administration is necessary before these drugs affect the gastrointestinal tract.
Torpet50 has reviewed oral side effects of cardiovascular drugs in humans. The list of possible lesions is impressive, as are the number of drugs associated with lesions, which suggests that the oral mucosa (at least in humans) is sensitive to the effects of many drugs. Lesions include taste disturbances (diuretics, angiotensin-converting enzyme inhibitors), xerostomia (e.g., alpha-agonists and beta or calcium channel blockers, angiotensin-converting enzyme inhibitors), gingival overgrowth (calcium and sodium channel blockers) and ulcerations, as well as a number of syndromes associated with cutaneous lesions indicative of drug allergies, including angioedema (many).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


