CHAPTER 62 Disseminated Intravascular Coagulation
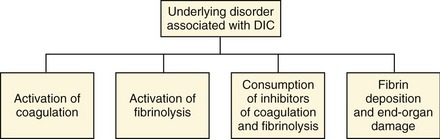
Blood normally circulates throughout the body in a fluid state. The body’s hemostatic system, composed of the coagulation cascade, platelets, vascular endothelium, and the fibrinolytic system, is responsible for maintaining the fluidity of blood while repairing vascular injuries. The coagulation cascade is activated during even minor states of disease or trauma. Normally, coagulation cascade activity is localized to the site of injury by natural anticoagulants and is in equilibrium with the fibrinolytic system. When there is sufficiently severe or prolonged activation, coagulation may occur unchecked and widespread microvascular thrombosis may occur, resulting in an often fatal condition known as disseminated intravascular coagulation (DIC). DIC has been defined as a disorder of systemic thrombosis and hemorrhage associated with underlying clinical situations and laboratory evidence of procoagulant activation, fibrinolytic activation, inhibitor consumption, and end-organ damage (Figure 62-1).1
ETIOLOGY
DIC is a disorder characterized in all species by the uncontrolled activation of the coagulation and fibrinolytic systems, resulting in widespread microvascular thrombosis and multiple organ dysfunction.2 DIC always occurs in association with an underlying condition that triggers the coagulation cascade. The most common causes of DIC in human beings are sepsis or severe inflammation/infection, neoplasia including both hemic and solid tumors, trauma, burns, and organ destruction including pancreatitis.1 In dogs, common underlying diseases include neoplasia (hemangiosarcoma, lymphoma, and other neoplasms), hepatic disease, sepsis, immune-mediated hemolytic anemia, renal disease, pancreatitis, trauma, immune-mediated thrombocytopenia, heat stroke, nonseptic peritonitis, gastric dilatation-volvulus, and benign splenic disease.3,4
A retrospective study published in 1995 evaluating hemostatic disorders in cats identified 21 patients fulfilling diagnostic criteria for DIC. The most common associated disorders were neoplasia, hepatic disease, and feline infectious peritonitis.5 Thomas et al identified 10 cats meeting criteria for DIC, with systemic infection, hepatic disease, and neoplasia as the most common underlying disorders.6 In a recent retrospective study of DIC in cats, the most common associated underlying disorders were neoplasia, pancreatitis, sepsis, and other infectious diseases (Table 62-1).7
Table 62-1 Final Diagnoses in Cats with Disseminated Intravascular Coagulation
| System or Category | Number Affected | Disorder (Number of Cats) |
|---|---|---|
| Neoplasia | 19 | Lymphoma (8), biliary adenocarcinoma (2), hepatocellular carcinoma (1), mastocytosis in spleen and bone marrow (1), metastatic pulmonary adenocarcinoma (1), multiple myeloma (1), metastatic carcinoma (1), pancreatic adenocarcinoma (1), myeloproliferative disease (not further characterized) with secondary IMHA (1), intrascapular fibrosarcoma (1), metastatic anaplastic neoplasia—not further characterized (1) |
| Pancreatic disease | 12 | Pancreatitis (12) |
| Sepsis | 9 | Cutaneous abscess/cellulitis (4), bacterial peritonitis (3), peritonitis secondary to intraabdominal neoplasia (2) |
| Infectious disease | 6 | Toxoplasmosis (2), yeast septicemia (1), feline panleukopenia virus (1), feline infectious peritonitis (1), feline leukemia virus (1) |
| Urinary system | 4 | Pyelonephritis (2), glomerular and renal medullary amyloidosis (1), uroabdomen (1) |
| Trauma/tissue necrosis | 2 | Trauma—fell four stories (1), tissue necrosis secondary to biliary cystadenoma (1) |
| Other | 3 | Immune-mediated hemolytic anemia (2), vaccine reaction (1) |
From Estrin MA, Wehausen CE, Jessen CR, et al: Disseminated intravascular coagulation in cats, J Vet Intern Med 20:1334, 2006.
EPIDEMIOLOGY
Because DIC is always secondary to a variety of underlying disease processes, all ages and breeds of cats are susceptible to developing the problem. From a veterinary teaching hospital population of 86 cats who fulfilled laboratory criteria for DIC (microscopic evidence of intravascular fibrin deposition or thrombosis in more than one organ on necropsy evaluation and/or three or more of the following: prolonged prothrombin time [PT], prolonged activated partial thromboplastin time [aPTT], thrombocytopenia, hypofibrinogenemia, and elevated fibrin/fibrinogen degradation products [FDPs]), the median age was 9 years with a range of 7 weeks to 23 years. Forty-four (51.1 per cent) of the cats were neutered males, one (1.1 per cent) cat was an intact male, 40 (46.5 per cent) cats were spayed females, and one cat (1.1 per cent) was an intact female. The median body weight was 4.5 kg (n = 84, range 0.76 to 9 kg). Sixty (69.8 per cent) of the cats were domestic shorthairs, while the remaining consisted of 15 domestic longhairs, two Siamese, and one of each of the following breeds: Persian, Himalayan, Abyssinian, Devon Rex, Birman, Ragdoll, British Blue, Norwegian Forest Cat, and a domestic mediumhair cat. Of those cats for whom data were available, 50 (58.1 per cent) cats lived indoors exclusively and 23 (26.7 per cent) cats lived both indoors and outdoors; none of the cats lived outdoors exclusively.8 The actual prevalence of DIC in the feline population is unknown.
COAGULATION IN THE NORMAL STATE
PHYSIOLOGY OF COAGULATION AND FIBRINOLYSIS
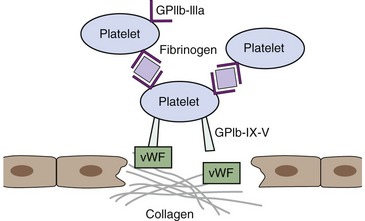
The process of hemostasis is a result of a complex series of interactions among the endothelium, platelets, and the coagulation cascade. In health, hemostasis is limited to the site of vascular injury by natural inhibitors of coagulation and the fibrinolytic system. The endothelium normally is resistant to thrombus formation, and the endothelial surface contains several modulators of coagulation, including a negative surface charge, thrombomodulin, heparan sulfate, and a protein C receptor.9 In states of vascular injury, circulating platelets are exposed to subendothelial connective tissue and von Willebrand factor (vWF). Platelets then bind to collagen and vWF via surface glycoprotein receptors and become activated, secreting molecules including thromboxane A2 and adenosine diphosphate (ADP) that activate locally circulating platelets. The activated platelets that cover the disrupted endothelium adhere via glycoprotein IIb/IIIa receptor binding to fibrinogen, which serves as a bridge to other activated platelets and leads to formation of a platelet aggregate.9 Receptors for activated coagulation factors are exposed on the platelet surface in states of activation (Figure 62-2).
The coagulation cascade was described previously as two separate pathways, intrinsic and extrinsic, both of which activated a common pathway. It is now considered as a series of interdependent enzymatic reactions involving coagulation factors, anticoagulant factors, and fibrinolytic factors (Figure 62-3). Coagulation factors circulate as inactive enzymes (prozymogens).
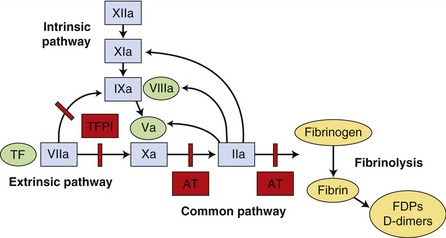
Tissue factor (TF) is an intrinsic membrane protein present on fibroblasts, neoplastic cells, and most other cell types not normally in contact with blood. Certain stimuli, such as cytokines interleukin-1 (IL-1) and tumor necrosis factor (TNF), can induce monocytes and endothelial cells to synthesize TF.9 Factor VII is capable of autoactivation, with activated factor VII (factor VIIa) normally present as 1 per cent of factor VII concentration. Factor VII and factor VIIa bind readily to TF, forming a complex on the cell surface. The TF-VIIa complex is capable of activating additional factor VII bound to TF. The TF-VIIa complex also activates factor IX and factor X, resulting in membrane-bound factor IXa and factor Xa. Factor IX also is activated by factor XIa, a protein in the contact system that is activated by thrombin and by factor XIIa. Factor XII is part of the contact system, which plays a role in fibrinolysis, inflammation, complement activation, and kinin formation but not in the initiation of coagulation.9 Activation of factor IX by factor XIa is thought to be less important in vivo than TF-VIIa activation because deficiency of factor XI results in a clinically mild hemostatic disorder in affected individuals.10 Similarly, a deficiency of factor XII and its cofactor, high-molecular-weight kininogen, does not result in a hemorrhagic tendency. Factor VIII, which circulates in plasma bound to vWF, can be activated by factor Xa or thrombin. Factor VIIIa serves as a cofactor for factor IXa and the resultant “Xase” complex is the most potent activator of factor X. Factor Xa converts prothrombin to thrombin in a slow reaction. The presence of factor Va helps form a prothrombinase complex on the cell surface and accelerates the formation of thrombin markedly. Factor V can be activated by both factor Xa and thrombin. Thrombin cleaves fibrinogen, forming a fibrin monomer. Fibrin monomers polymerize spontaneously and then are cross-linked covalently to form a stable fibrin clot by factor XIIIa, which is activated by thrombin.
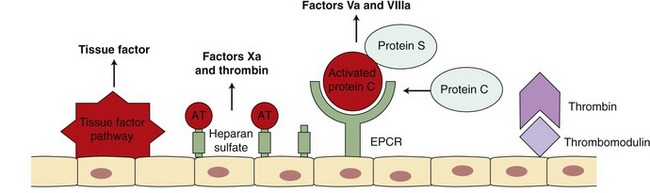
Procoagulant activity of the coagulation factors is balanced by inhibitors of coagulation (Figure 62-4). Several physiological mechanisms inhibit coagulation. TF pathway inhibitor regulates the activation of factors IX and X by the TF-VIIa complex. Antithrombin (AT) inhibits thrombin, factor Xa, and factor IXa.11 The activity of AT is enhanced greatly by heparin. Factor Xa also is inhibited by Z protease inhibitor and protein Z.9 Thrombin bound to thrombomodulin on the endothelial surface is not capable of activating factors V and VIII but does activate protein C. Activated protein C (APC), along with its cofactor protein S, inactivates factors Va and VIIIa.11 This serves as an important regulatory pathway because the inactivated factors V and VIII are no longer able to participate in the coagulation cascade.
The fibrinolytic system solubilizes fibrin clots, maintaining the balance between fibrin clot formation and dissolution. Thrombin stimulates endothelial cells to release tissue-type plasminogen activator (tPA).11 Plasminogen is activated to plasmin by tPA when both are bound to fibrin. Plasmin splits fibrinogen and fibrin to produce fibrin degradation products (FDPs). These products are themselves anticoagulants, inhibiting thrombin and fibrin polymerization.11 Cross-linked fibrin is relatively more resistant to plasmin degradation. Urokinase plasminogen activator acts similarly to tPA and is formed by activation of prourokinase. Prourokinase bound to its receptor on activated endothelial cells is activated by plasmin, by autoactivation, and by kallikrein, a component of the contact system.11 Urokinase then cleaves plasminogen to form plasmin. The fibrinolytic system is regulated by α-2 antiplasmin, plasminogen activator inhibitor-1 (PAI-1), and thrombin-activated fibrinolysis inhibitor.
DISRUPTION OF NORMAL PROCOAGULANT, ANTICOAGULANT, AND FIBRINOLYTIC BALANCE
DIC results from excess systemic generation of thrombin, which leads to a disruption of balance between coagulation and fibrinolysis. Major causes of marked activation of coagulation sufficient to lead to DIC include endothelial injury, tissue injury, and monocyte activation. TF is the key event in most cases of initiation of DIC, and studies in animal models of Escherichia coli endotoxemia showed that inhibition of the TF pathway eliminated thrombin production.12 Systemic generation of thrombin has been demonstrated to be mediated by the extrinsic pathway of coagulation via TF and factor VIIa.12 TF is expressed on activated mononuclear cells, endothelial cells, and neoplastic cells.2 Endotoxin, IL-1, and TNF induce TF synthesis and expression on endothelial cells.13 TNFα transcription is stimulated by the TF-VIIa complex, leading to increased TF synthesis and resulting in a positive feedback loop.13 TNFα stimulates the release of other cytokines, including IL-1 and IL-6.13 IL-6 contributes to systemic thrombin generation by causing TF expression on mononuclear cells.
ABNORMALITIES OF THE FIBRINOLYTIC SYSTEM
As a result of widespread thrombin production and subsequent release of tPA, plasminogen is converted to plasmin and increased levels of plasmin circulate systemically during DIC, acting on fibrin and fibrinogen to produce FDPs X, Y, D, and E.4 Plasmin also splits cross-linked fibrin, creating the D-dimer fragment. Other enzymes degraded by plasmin include factors V, VIII, IX, and XI, which results in decreased concentrations of these clotting factors.1 The FDPs in circulation interfere with fibrin polymerization, leading to hemorrhage. FDPs sometimes are referred to as fibrin split products (FSPs) or fibrin-related antigens.14 FDPs activate macrophages and monocytes to release IL-1, IL-6, and PAI-1, a potent inhibitor of fibrinolysis.1 During peak activation of coagulation, endotoxin, IL-1, and TNF increase PAI-1 synthesis and reduce thrombomodulin expression, thus inhibiting fibrinolysis and favoring coagulation.2
INHIBITOR CONSUMPTION
AT and protein C concentrations decrease in patients with DIC. AT levels are reduced in DIC because of consumption, reduced synthesis, and degradation by neutrophil elastase.2 Protein C activity is compromised as a result of reduced synthesis, reduced thrombomodulin expression on endothelial cells, and binding of cofactor protein S to C4b-binding protein.2 TF pathway inhibitor function also is insufficient in states of DIC.2
END-ORGAN DAMAGE
Systemic circulation of thrombin results in the splitting of fibrinogen to form fibrinopeptides A and B and the fibrin monomer.1 Fibrin monomers polymerize spontaneously and factor XIII, activated by thrombin, stabilizes the cross-linked fibrin, forming a clot that contributes to tissue ischemia. IL-1 and IL-6 cause vascular endothelial disruption and thus contribute to end-organ damage.15 Thrombin-induced release of endothelin from endothelial cells leads to vasoconstriction and vasospasm, predisposing to thrombus formation and vascular occlusion.1
CLINICAL FEATURES
The clinical signs in cats with DIC are nonspecific and often reflect the particular underlying disorder. A review of 86 hospitalized cats with DIC revealed that depression and lethargy were the primary complaint, followed by anorexia, weakness, vomiting, dyspnea, and diarrhea. Common physical examination abnormalities included hypothermia, weak femoral pulses, tachycardia, hyperthermia, a palpable abdominal mass, cardiac murmur, bradycardia, and presence of a gallop rhythm.8
Physical examination findings compatible with a coagulation disorder are uncommon despite frequent alterations in laboratory tests of coagulation. Lack of spontaneous hemorrhage is common in cats with abnormal laboratory tests of coagulation. Peterson et al found evidence of spontaneous bleeding in only one of 21 cats with DIC.5 In a study by Estrin et al,7 hemorrhage was noted in only 14 of 86 (16.3 per cent) cats. Petechiae or ecchymoses were noted in six of 86 (seven per cent) cats. Other sources of hemorrhage consisted of hemoabdomen (5), rectal bleeding (2), epistaxis (3), hemothorax (3), and hematuria (1) (Figure 62-5). One cat had both rectal bleeding and epistaxis, while another cat had both a hemothorax and hemoabdomen. The three cats with the lowest platelet counts (1,000 to 13,000/µL) exhibited hemorrhage. One cat had epistaxis and petechiae, one had rectal bleeding and petechiae, and one developed hemoabdomen. Hemorrhage was noted in five of 12 cats with a prothrombin time (PT) of 120 seconds or higher (reference range 7.4 to 12.8 seconds) and in six of 14 cats with an activated partial thromboplastin time (aPTT) of 120 seconds or higher (reference range 11.1 to 16.4 seconds).7

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


