Chapter 2 DISORDERS OF GROWTH HORMONE
BIOSYNTHESIS OF GROWTH HORMONE AND IGF-I
Growth hormone (GH; somatotropin) is a single-chain polypeptide with a molecular weight of approximately 22,000 daltons (Thorner et al, 1998). The amino acid sequence of canine GH has been elucidated and is identical to that of porcine GH (Mol and Rijnberk, 1997; Secchi et al, 2001). GH shares amino acid sequence homology with prolactin and placental lactogen, suggesting that these hormones evolved from a single ancestral gene. GH is synthesized and secreted by the somatotrophs of the anterior pituitary. GH’s larger precursor peptide, PreGH, is also secreted but has no known physiologic significance (Aron et al, 2001). Secretion of GH occurs in a pulsatile ultradian fashion, with pulse intervals of 4 to 6 hours (French et al, 1987; Conzemius et al, 1998). In humans, plasma GH concentrations increase from birth onward, reach a maximum in late adolescence, and decline during adulthood. In contrast, in a study by Nap and colleagues (1993), mean plasma GH concentrations declined between 7 and 27 weeks of age (13.7 ± 2.2 versus 0.7 ± 0.4 μg/L, respectively) in Great Dane puppies. The pulsatile secretion pattern of GH also changes according to the phase of the estrus cycle in healthy bitches, with higher baseline GH secretion and less GH secreted in pulses during stages involving a high plasma progesterone concentration (Kooistra et al, 2000a). The pulse height of GH decreases with age in humans, cattle, and domestic fowl and is presumed to also decrease with age in other species, including dogs and cats (Mol and Rijnberk, 1997). High- and low-affinity GH-binding proteins (GHBPs) exist in plasma in humans and have been reported in dogs (Maxwell et al, 2000). GHBPs are believed to play a significant role in modulating the activity of growth hormone (Styne, 2001). The high-affinity GHBP is identical to the amino acid sequence of the extracellular domain of the GH receptor (Thorner et al, 1998). Protein-bound GH is metabolized differently, persists longer in plasma, and has a limited volume of distribution compared with monomeric GH, suggesting that protein binding may enhance the biologic activity of GH.
In the late 1950s researchers identified a “sulfation factor,” which promoted growth and was controlled by GH, and a serum fraction, which had insulin-like activity that could not be abolished by antiinsulin antibodies. In the 1970s these factors were termed somatomedins (because they mediate the action of somatotropin) and insulin-like growth factors, respectively. Somatomedin C and insulin-like growth factor-I (IGF-I) were subsequently shown to be the same molecule. Most of the growth-promoting effects of GH are mediated by IGF-I. IGF-I has an approximately 50% homology of structure with proinsulin and insulin, and the IGF-I cell membrane receptor resembles the insulin receptor in its structure. IGF-I binds to insulin receptors and promotes glucose utilization, enhances tissue sensitivity to insulin, and decreases the blood glucose concentration. IGF-I is produced in most tissues and appears to be exported to neighboring cells to act upon them in a paracrine manner (Yakar et al, 1999; Styne, 2001). As such, serum IGF-I concentrations may not reflect the most significant actions of this growth factor. The liver is a major site of IGF-I synthesis, and much of the circulating IGF-I is believed to be derived from the liver. In humans, serum IGF-I concentrations vary in liver disease with the extent of liver destruction.
Secretion of IGF-I is under the direct control of GH. Serum IGF-I concentrations are low in GH deficiency and increased in acromegaly. The biologic effects of circulating IGF-I are modulated by several different IGF-binding proteins (IGFBPs), with IGFBP-3 being the most abundant in dogs and cats (Maxwell et al, 1998; Lewitt et al, 2000). In dogs, IGF-I binds primarily with IGFBP-3 and an acid-labile subunit (ALS) to form a ternary complex that does not cross the vascular endothelium, that prolongs the plasma half-life of IGF-I, and that functions as a storage pool for IGF-I (Maxwell et al, 2000). In one study, ALS concentrations were low in cats, compared with other species, and this limited the formation of IGFBP-3 ternary complexes (Lewitt et al, 2000). Interestingly, ALS concentrations were increased in diabetic cats, and the elevated levels, by promoting ternary complex formation, may have lead to the increase seen in serum IGF-I concentrations in the diabetic cats compared with healthy cats; this finding may have implications for the use of IGF-I as a diagnostic aid for feline acromegaly (see page 76).
IGFBP-1 is the only acute regulator of IGF-I bioavailability (Lee et al, 1997). The primary role of IGFBP-1 is the acute binding of IGF-I in the circulation; the greater the concentration of IGFBP-1, the lower the concentration of free IGF-I and vice versa. GH decreases IGFBP-1 concentrations in plasma. Results of in vitro and in vivo studies have shown that IGFBP-1 inhibits growth and differentiation stimulated by IGF-I and counteracts the insulin-like hypoglycemic activity of IGF-I (Ferry et al, 1999). Circulating IGF-I and its interaction with IGFBP-1 are believed to be important determinants of glucose homeostasis, providing an important link between nutrition and growth (Sandhu et al, 2002). For example, consumption of a meal stimulates ghrelin secretion, which stimulates GH secretion, which decreases IGFBP-1 and increases the amount of free IGF-I available for binding to insulin receptors and promoting glucose utilization. Insulin suppresses IGFBP-1, and studies have documented decreased IGFBP-1 levels in humans with obesity and type 2 diabetes mellitus; these findings suggest that measurement of IGFBP-1 may be useful as a marker for insulin resistance. Finally, some disease states increase the levels of IGFBP-1, which can have a deleterious effect on the diagnostic usefulness of IGF-I as a marker for GH excess. Renal failure and some solid tumors have been shown to increase the concentration of IGFBP-1 in humans, causing an increase in plasma total IGF-I concentrations by as much as 30% to 50%.
METABOLIC ACTIONS OF GROWTH HORMONE
GH is described as anabolic, lipolytic, and diabetogenic, concepts derived primarily from in vitro studies of isolated tissue and in vivo studies involving administration of pharmacologic amounts of GH. The primary function of growth hormone is promotion of linear growth (Conzemius et al, 1998). Most of the growth-promoting effects are mediated by IGF-I (Fig. 2-1). A dual-effector model of GH action has been proposed in which GH first stimulates prechondrocytes in the epiphyseal growth plate to undergo differentiation and stimulates the expression of IGF-I in hepatocytes and chondrocytes; IGF-I then acts as a mitogen to stimulate clonal growth of the differentiated cells (Isaksson et al, 1987; Scheven and Hamilton, 1991). In this way locally produced IGF-I, under GH regulation, contributes to the stimulatory effects of GH, particularly the enhancement of longitudinal growth.
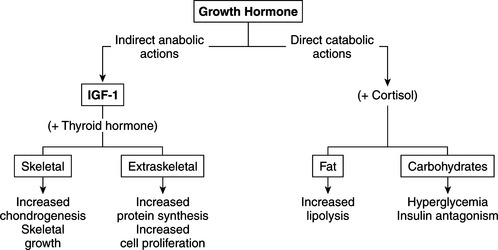
FIGURE 2-1 The anabolic and catabolic actions of growth hormone.
(Modified from Underwood LE, Van Wyk JJ: In Wilson J, Foster D (eds): Williams Textbook of Endocrinology, 7th ed. Philadelphia, WB Saunders, 1985, p 155.)
Differences in adult body size of dog breeds is preceded by differences in GH release at a young age and not by differences in circulating IGF-I concentrations. In one study, young Great Danes and young Beagles both experienced a period of high GH release, but this period persisted much longer in Great Danes (Favier et al, 2001). There was no difference in circulating IGF-I concentrations between the two groups of young dogs. The authors speculate that the longer period of increased GH secretion in the Great Danes may have been caused by delayed maturation of the inhibitory influences of somatostatin on GH release. In contrast, during adulthood similar GH concentrations are found in the plasma of dog breeds of widely differing body sizes, but the serum IGF-I levels are quite different and positively correlated with body size (Eigenmann, 1987). Smaller breeds have lower concentrations of IGF-I than larger breeds. In one study, the mean IGF-I concentrations were 36 ± 27 ng/ml in Cocker Spaniels; 87 ± 33 ng/ml in Beagles; 117 ± 34 ng/ml in Keeshonds; and 280 ± 23 ng/ml in German Shepherd dogs (Eigenmann et al, 1984b). In another study that compared IGF-I levels in different-sized dogs within the same breed (i.e., Toy, Miniature, and Standard Poodles), a linear correlation between body size and circulating IGF-I levels was established (Eigenmann et al, 1984a). All of these dogs secreted similar normal amounts of GH in response to provocative stimulation.
Another important anabolic action of GH is stimulation of protein synthesis, an action mediated by IGF-I (see Fig. 2-1). Cellular uptake of amino acids is enhanced; transcription and translation of mRNA is accelerated; and enzymatic steps necessary for protein synthesis are activated (Daughaday, 1985). DNA synthesis is stimulated in a variety of cell types, including fibroblasts, chondrocytes, muscle, and hematopoietic and hepatic tissue (Van Wyk et al, 1981). These actions result in the stimulation of cell proliferation or in cellular differentiation without obligatory proliferation. In all responding tissues, the production of cell products is characteristic of each target tissue.
GH tends to decrease protein catabolism by directly promoting lipolysis and enhancing the conversion of free fatty acids to acetyl coenzyme A (acetyl-CoA), from which energy is derived (see Fig. 2-1; Aron et al, 2001). GH also has a direct effect on carbohydrate metabolism. In excess, it decreases carbohydrate utilization and impairs glucose uptake into cells. GH-induced insulin resistance appears to be due to a postreceptor impairment in insulin action (Bratusch-Marrain et al, 1982; Rosenfeld et al, 1982). The net effect is promotion of hyperglycemia, carbohydrate intolerance, and, with sustained increases in plasma GH, the development of diabetes mellitus, which quickly becomes resistant to insulin treatment.
REGULATION OF GROWTH HORMONE SECRETION
In many domestic species, GH is secreted from the pituitary gland in a pulsatile manner that is under the dual regulation of hypothalamic GH-releasing hormone (GHRH; somatocrinin) and GH release-inhibiting factor (somatostatin) (Fig. 2-2). The two factors act in concert to precisely control GH secretion so as to maintain homeostasis in the growing body from fetal life through adolescence. These positive and negative regulating stimuli persist in adults as well.
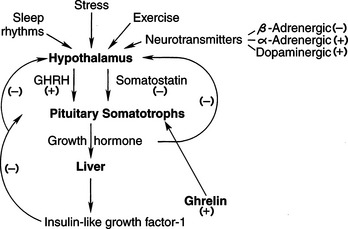
GH-RELEASING HORMONE.
GHRH stimulates both GH synthesis and secretion. GHRH causes a prompt increase in blood GH levels, followed by a rapid return of GH to basal concentrations (Thorner et al, 1983). The pulsatile increases in GH primarily reflect the pulsatile secretion of GHRH from the hypothalamus (Thorner et al, 1998). Sustained infusions of GHRH over several hours result in an initial rise and then a progressive decrease in plasma GH concentrations, suggesting that GHRH depends on pulsatile secretion for its physiologic effect (Reichlin, 1998). The effects of GHRH are almost completely specific for GH, although a minimal increase in plasma prolactin may also occur.
SOMATOSTATIN.
Somatostatin denotes a family of peptides, including somatostatin-14, which is a cyclic peptide containing 14 amino acids, and the N-terminal-extended somatostatin-28, which is a cyclic peptide containing 28 amino acids. Somatostatin-14 is identical with the terminal 14 amino acids of somatostatin-28. Somatostatin-14 is found predominately in the central nervous system (CNS), including the hypothalamus, and somatostatin-28 is found predominately in the gastrointestinal tract, including the pancreas (Reichlin, 1998). As a pituitary regulator, somatostatin is a potent inhibitor of GH secretion. Somatostatin secretion is increased by elevated levels of GH and IGF-I. Somatostatin appears to determine the timing and amplitude of GH pulses but has no effect on GH synthesis (Thorner et al, 1998). Somatostatin inhibits secretion of thyroid-stimulating hormone (TSH) and, under certain conditions, secretion of prolactin and adrenocorticotropic hormone (ACTH). It also has nonpituitary roles, including activity as a neuromodulator in the central and peripheral nervous systems and as a gut and pancreatic regulatory peptide (Table 2-1; Reichlin, 1998). Somatostatin blocks hormone release in many endocrine-secreting tumors, including insulinomas, glucagonomas, vasoactive intestinal polypeptide-secreting tumors (VIPomas), carcinoid tumors, and some gastrinomas (see Chapters 14 and 15). In humans, long-acting analogs of somatostatin have been used therapeutically in the management of GH excess and functioning neuroendocrine tumors (Reichlin, 1998).
TABLE 2-1 BIOLOGIC ACTIONS OF SOMATOSTATIN OUTSIDE THE CENTRAL NERVOUS SYSTEM
| Inhibition of Hormone Secretion | Inhibition of Gastrointestinal Actions |
|---|---|
TSH, thyrotropin; GH, growth hormone; AVP, arginine vasopressin; VIP, vasoactive intestinal polypeptide.
Modified from Reichlin S: In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed. Philadelphia, WB Saunders Co, 1998, p 191.
STIMULI AFFECTING GH-REGULATING FACTORS.
The influence of GHRH and somatostatin on GH secretion is tightly regulated by an integrated system of neural, metabolic, and hormonal factors, including GH itself (Tables 2-2 and 2-3) (Buonomo and Baile, 1990). Most of these stimuli exert their effects on GH secretion by influencing hypothalamic secretion of GHRH or somatostatin. The predominant influence of the hypothalamus on GH secretion is stimulatory, as evidenced by the fact that damage to the hypothalamic-pituitary connection is followed by inhibition of both basal and induced GH release (Reichlin, 1998). One exception is ghrelin, a recently discovered hormone that is produced primarily in the stomach and to a lesser extent in the hypothalamus. Ghrelin is secreted after consumption of a meal; its primary actions are to decrease gastric emptying and promote satiety. Ghrelin also stimulates GH secretion, which in turn increases IGF-I concentrations. IGF-I promotes glucose utilization by binding to insulin receptors in tissues. Growth hormone also decreases IGFBP-1 concentrations, resulting in increased free IGF-I concentrations. As mentioned previously, the primary role of IGFBP-1 is the binding of IGF-I, thereby decreasing the circulating concentration of IGF-I.
TABLE 2-2 FACTORS THAT STIMULATE GH SECRETION IN PRIMATES
Modified from Reichlin S: In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed. Philadelphia, WB Saunders Co, 1998, p 210. (source)
TABLE 2-3 FACTORS THAT INHIBIT GH SECRETION IN PRIMATES
Modified from Reichlin S: In Wilson JD, Foster DW, Kronenberg HM, Larsen PR (eds): Williams Textbook of Endocrinology, 9th ed. Philadelphia, WB Saunders Co, 1998, p 210. (source)
CONGENITAL HYPOSOMATOTROPISM: PITUITARY DWARFISM
Etiology
Pituitary dwarfism results from a congenital deficiency of GH. GH deficiency may occur as an isolated deficiency or in combination with deficiencies in other hormones of the anterior lobe of the pituitary gland (i.e., TSH, prolactin, ACTH, follicle-stimulating hormone [FSH], and luteinizing hormone [LH]). In dogs and probably cats, dwarfism is most commonly associated with pressure atrophy of the anterior lobe of the pituitary gland caused by cystic enlargement of the residual craniopharyngeal duct (i.e., Rathke’s cleft) and with pituitary hypoplasia resulting from primary failure of differentiation of the craniopharyngeal ectoderm of Rathke’s pouch into a normal anterior lobe.
PITUITARY CYST.
Pituitary dwarfism may result from congenital cystic distention or persistence of the “intrasellar” portion of the craniopharyngeal duct (Rathke’s pouch). The intrasellar portion is found within the sella turcica, the region of the skull “encasing” the pituitary. During embryonic development, the anterior pituitary (adenohypophysis) arises from Rathke’s pouch, an evagination of the posterior wall of the pharynx. The portion of the pouch that comes in contact with the neurohypophysis differentiates into the pars intermedia of the anterior pituitary, and the anterior wall of the pouch differentiates into the pars distalis of the anterior pituitary. Lateral proliferations from the embryonic pouch extend around the infundibular stem to form the pars tuberalis (pituitary stalk). In the dog and cat, the distal and intermediate parts of the anterior pituitary are separated by the residual hypophyseal cleft, which is the residual lumen of Rathke’s pouch. The stalk of Rathke’s pouch is attenuated in the embryo to form the craniopharyngeal duct. This duct should disappear, although either or both ends of it commonly persist as vestiges in dogs. Persistence of the intrasellar portions of the duct is especially common in brachycephalic breeds. The degree of persistence and consequences varies. The remnants may become cystic; may be single or multiple, unilocular or multilocular; and frequently contain mucin produced by the epithelium that lines the cyst. The cysts are usually microscopic and clinically insignificant but sometimes are larger in volume than a normal pituitary gland (Fig. 2-3). Large cysts may cause pressure atrophy of pituitary cells.
Pituitary cysts are relatively common in the dog, yet dwarfism is rare. Also, dwarfism is most often encountered in the German Shepherd dog, and German Shepherd dwarfs have been identified with only a very small pituitary cyst. For these reasons, the theory that cystic distention of the craniopharyngeal duct is the primary cause of destruction of the adenohypophysis has been questioned. An alternative hypothesis suggests a primary failure in the differentiation of the adenohypophyseal cells into normal tropic hormone-secreting cells (Kooistra et al, 2000b).
PITUITARY HYPOPLASIA.
The development of the anterior lobe of the pituitary gland is characterized by the expression of a series of homeodomain transcription factors, which are required for normal growth of the pituitary primordium (Treier et al, 1998; Watkins-Chow and Camper, 1998). After proliferation, different cell phenotypes arise in a distinct temporal fashion and undergo a highly selective determination and differentiation (Simmons et al, 1990; Voss and Rosenfeld, 1992). The corticotropic cell is the first distinct cell type that differentiates from the pituitary stem cell (Sheng et al, 1997). Ultimately, the mature pituitary anterior lobe is composed of five distinct endocrine cell types that are easily distinguished by the hormones they secrete. Any defect in the organogenesis of the pituitary gland may result in an isolated or combined pituitary hormone deficiency.
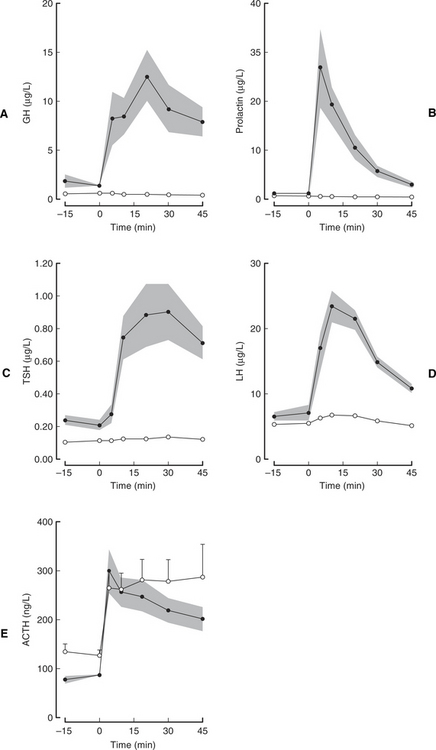
Pituitary dwarfism is encountered most often as a simple, autosomal recessive inherited abnormality in the German Shepherd dog (Kooistra et al, 2000b). Inherited pituitary dwarfism may be due to isolated GH deficiency or may be part of a combined anterior pituitary hormone deficiency syndrome. Concurrent deficiency in TSH and prolactin are most commonly identified in German Shepherd dwarfs; ACTH secretion is preserved (Fig. 2-4; Hamann et al, 1999; Kooistra et al, 2000b). Kooistra and colleagues (2000b) hypothesize that the disorder in German Shepherd dogs is caused by a mutation in a developmental transcription factor that precludes effective expansion of a pituitary stem cell after the differentiation of the corticotropic cells that produce ACTH. Mutations in the genes encoding for transcription factors Pit-1 or the Prophet of Pit-1 (Prop-1) cause combined pituitary anterior lobe hormone deficiencies in humans and mice (Pellegrini-Bouiller et al, 1996; Fofanova et al, 1998; Wu et al, 1998). Unfortunately, a mutation in the gene encoding transcription factors Pit-1 and Prop-1 was not identified in German Shepherd dwarfs, which indicates that another homeodomain gene is affected in dwarf German Shepherd dogs (Lantinga-van Leeuwen et al, 2000a and 2000b). Kooistra and colleagues (2000b) speculate that the underlying defect may involve a mutation that precludes effective expansion of a pituitary stem cell. Because ACTH secretion remains intact, a combined pituitary hormone deficiency in German Shepherd dwarfs would most likely be caused by a mutation in a developmental transcription factor that precludes effective expansion of a pituitary stem cell after the differentiation of the corticotropic cells. Such a factor would be analogous to the pituitary transcription factors Pit-1 and Prop-1 but would act at an earlier stage of pituitary gland development (Kooistra et al, 2000b).
The cystic changes involving the pituitary gland in dogs with congenital GH deficiency may be the result of primary failure of differentiation of the anterior lobe of the pituitary gland rather than the cause of the GH deficiency. Undifferentiated adenohypophyseal cells may secrete osmotically active proteinaceous material into the craniopharyngeal duct (Eigenmann, 1983). Analysis of cystic fluid obtained from rats has shown that the osmotically active proteinaceous material could attract water and lead to cystic changes, which is compatible with the observation that the cysts enlarge with time (Benjamin, 1981; Kooistra et al, 1998).
GH INSENSITIVITY SYNDROMES.
Pituitary dwarfism may develop from insensitivity to GH. The prototype of GH insensitivity is the syndrome of Laron-type dwarfism in human beings. Circulating levels of GH are increased in people with Laron-type dwarfism, but IGF-I levels are deficient. This disorder is due to inactivating mutations in the gene for the GH receptor, which results in absent or defective GH receptors and partial to complete GH insensitivity (Rosenfeld et al, 1994; Goddard et al, 1995). GH insensitivity can also result from an abnormality in the structure of GH (Takahashi et al, 1996), abnormalities of GH signal transduction (postreceptor defects), primary defects of synthesis of IGF-I, or lack of target tissue response to IGF-I (Lanes et al, 1980; Takahashi et al, 1996; Reiter and Rosenfeld, 1998).
Similar subcategories of dwarfism in dogs or cats have not yet been convincingly described. To date, all cases of pituitary dwarfism in the dog in which either GH or IGF-I concentrations were evaluated have revealed low to undetectable GH concentrations and low IGF-I concentrations. There has been no response of GH to stimulation tests (Rijnberk et al, 1993). Two dwarf German Shepherd dogs have been described with low IGF-I activity and histologically normal pituitary glands. Secondary GH deficiency (i.e., abnormal GH structure or nonresponsive target tissue) was proposed as the cause for the dwarfism, making the assumption that circulating GH levels were normal. Unfortunately, plasma GH concentrations were not evaluated to substantiate the claims. In another study in a German Shepherd dog, similar arguments were made for secondary GH deficiency as the cause of the dwarfism (Muller-Peddinghaus et al, 1980). In this study, basal GH concentrations rather than the GH secretory capacity of the pituitary were assessed. Unfortunately, basal GH concentrations in normal and hypopituitary dogs may be similar, rendering this parameter inadequate for assessing GH secretory capacity (Eigenmann and Eigenmann, 1981a).
Interestingly, two German Shepherd dogs have been described that had delayed growth, resulting in an initial stunted appearance, that eventually resolved by 1 year of age (Randolph et al, 1990). Both dogs had normal basal serum GH and IGF-I concentrations, normal GH secretory response to xylazine administration, and normal adrenal gland and thyroid gland function. These authors speculated that delayed growth was due to mild hypopituitarism or intermittent GH neurosecretory dysfunction. It seems logical that a spectrum of severity, from mild to severe, might exist with hypopituitarism and that the exhibited clinical signs would depend on the severity of impaired GH secretion.
Pathophysiology
The clinical manifestations of pituitary dwarfism result from the deficiency in GH (hyposomatotropism) and the secondary deficiency in circulating IGF-I (Eigenmann et al, 1984c). In the immature animal, hyposomatotropism impairs linear growth, resulting in the development of short stature. Dogs with combined anterior pituitary hormone deficiencies usually lack TSH and prolactin in addition to GH. Lack of TSH secretion may contribute to abnormal body maturation and growth (see Chapter 3). Normal physical development depends on the presence of normal plasma thyroid hormone concentrations, which act synergistically with GH and IGF-I to promote chondrogenesis. Lack of prolactin secretion would prevent mammary gland enlargement and lactation in the postpartum bitch. Dogs with combined anterior pituitary hormone deficiencies may have impaired secretion of gonadotropins (LH and FSH), resulting in hypogonadism and infertility. Studies in German Shepherd dwarfs suggest that corticotroph function and ACTH secretion are maintained in dogs with combined pituitary hormone deficiencies (Kooistra et al, 2000b). Secondary adrenal insufficiency does not occur in most dogs with combined anterior pituitary hormone deficiencies. Clinical manifestations resulting from deficiency of neurohypophyseal hormones (i.e., vasopressin, oxytocin) are not apparent in pituitary dwarfs. If the neurohypophysis were damaged, vasopressin secretion would continue directly from the hypothalamus, thereby preventing diabetes insipidus. Because of concurrent hypogonadism, any deficiency in oxytocin would be clinically insignificant.
Signalment
Pituitary dwarfism occurs primarily in the German Shepherd dog, although the condition has been described in other breeds, including the Weimaraner, Spitz, Toy Pinscher, and Carnelian Bear dog. Pituitary dwarfism has also been described in cats. A simple autosomal recessive mode of inheritance has been reported for the German Shepherd dog and the Carnelian Bear dog. In one study, plasma IGF-I concentrations were evaluated in a group of German Shepherd dwarfs and their relatives; interestingly, in the clinically unaffected relatives, IGF-I concentrations were intermediate between those of the normal dogs and those of the dwarfs, suggesting a gene-dosage effect (Willeberg et al 1975). This pattern would be expected if the unaffected relatives were heterozygous, expressing approximately 50% of the trait compared with the fully expressed homozygous dwarfs. Unfortunately, determinations of the basal GH concentrations and the responsiveness of the somatotrophs to provocative testing were not performed.
Clinical signs
The most common clinical manifestations of pituitary dwarfism are a lack of growth (i.e., short stature), endocrine alopecia, and hyperpigmentation of the skin (Table 2-4). Affected animals are usually normal in size during the first 1 to 2 months of life, after which the rate of growth is slower than that of their littermates. By 3 to 4 months of age, affected dogs and cats are obviously “runts” of the litter and usually never attain full adult dimensions (Figs. 2-5 and 2-6). The initial normal growth is consistent with the concept that growth in early postnatal life in most animals and in humans proceeds for a certain period at a normal rate even in the absence of GH.
TABLE 2-4 CLINICAL SIGNS ASSOCIATED WITH PITUITARY DWARFISM

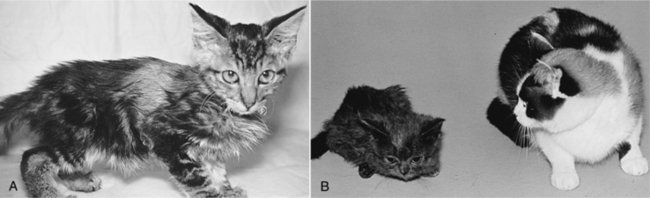
Dwarfs with an isolated GH deficiency typically maintain a normal body contour and body proportions as they age (i.e., proportionate dwarfism), whereas dwarfs with combined anterior pituitary hormone deficiencies (most notably TSH) may acquire a square or chunky contour typically associated with congenital hypothyroidism (i.e., disproportionate dwarfism) (see Fig. 2-6) (see Chapter 3, page 104). Closure of the epiphyseal growth plates is usually delayed in pituitary dwarfs, and the fontanelles of the skull may remain open. Dental eruption may be delayed, but dentition is usually normal.
The most notable dermatologic sign is retention of the lanugo, or secondary, hairs, with concurrent lack of the primary, or guard, hairs. As a result, the hair coat in a dwarf is initially soft and woolly. The lanugo hairs are easily epilated, and bilateral symmetric alopecia gradually develops. Initially, hair loss is confined to areas of wear, such as the neck (collar) and posterolateral aspects of the thighs (from sitting). Eventually the entire trunk, neck, and proximal limbs become alopecic, with primary hairs remaining only on the face and distal extremities.
Hypogonadism may also develop, although normal reproductive function has been reported in some pituitary dwarfs. In the male, testicular atrophy, azoospermia, and a flaccid penile sheath are typical; in the female, absence of estrus activity develops as a result of impaired secretion of pituitary gonadotropins. Most canine pituitary dwarfs retain a shrill, puppylike bark (Campbell, 1988). Most pituitary dwarfs remain alert and active when young. As they grow older, however, they may become progressively more listless, dull, and inactive. Inappetence may develop. These changes may reflect the various endocrine deficiencies associated with the condition or progressive expansion of a pituitary cyst or cysts.
Clinical pathology
The results of a complete blood count (CBC), biochemical panel, and urinalysis are usually normal in dogs and cats with pituitary dwarfism caused by isolated GH deficiency. A mild increase in renal parameters (urea nitrogen and creatinine) may develop secondary to maldevelopment of glomeruli or a functional decrease in the glomerular filtration rate (Feld and Hirschberg, 1996). Hypophosphatemia, hypoalbuminemia, and anemia have also been reported (Eigenmann, 1983). Additional clinicopathologic alterations may develop in pituitary dwarfs with combined anterior pituitary hormone deficiencies, primarily as a result of concurrent hypothyroidism (see Chapter 3).
Dermatohistopathology
The histopathologic alterations in the skin of dogs with hyposomatotropism are similar to those seen in many other endocrinopathies (Table 2-5). These skin alterations include varying degrees of orthokeratotic hyperkeratosis, follicular keratosis, follicular dilatation, follicular atrophy, telogenization of hair follicles, excessive trichilemmal keratinization, sebaceous gland atrophy, epidermal atrophy, epidermal melanosis, and dermal thinning (Scott et al, 2001). A highly suggestive finding of hyposomatotropism is the decreased amount and size of dermal elastin fibers. Elastin fibers are smaller, fragmented, and less numerous than normal. In dogs with concurrent secondary hypothyroidism, histopathologic findings may include vacuolated or hypertrophied arrector pili muscles.
TABLE 2-5 DERMATOHISTOPATHOLOGIC ALTERATIONS ASSOCIATED WITH ENDOCRINOPATHY-INDUCED ALOPECIA
| Abnormality | Specific Endocrine Disorder |
|---|---|
| Nonspecific Abnormalities Supporting an Endocrinopathy | |
| Orthokeratotic hyperkeratosis | — |
| Follicular keratosis | — |
| Follicular dilatation | — |
| Follicular atrophy | — |
| Predominance of telogen hair follicles | — |
| Sebaceous gland atrophy | — |
| Epidermal atrophy | — |
| Epidermal melanosis | — |
| Thin dermis | — |
| Dermal collagen atrophy | — |
| Abnormalities Suggestive of Specific Endocrine Disorder | |
| Decreased amount and size of dermal elastin fibers | Hyposomatotropism |
| Excessive trichilemmal keratinization (flame follicles) | Growth hormone- and castration-responsive dermatosis |
| Vacuolated and/or hypertrophied arrector pilae muscles | Hypothyroidism |
| Increased dermal mucin content | Hypothyroidism |
| Thick dermis | Hypothyroidism |
| Comedones | Hyperadrenocorticism |
| Calcinosis cutis | Hyperadrenocorticism |
| Absence of arrector pili muscles | Hyperadrenocorticism |
Endocrinologic evaluation
BASAL GH CONCENTRATIONS.
Reported normal basal GH concentrations range from 1.5 ± 1.2 ng/ml to 4.3 ± 1.1 ng/ml in dogs (Eigenmann, 1983) and 1.2 ± 1.0 ng/ml to 3.2 ± 0.7 ng/ml in cats (Eigenmann et al, 1984d; Peterson et al, 1990). Unfortunately, basal GH concentrations in dogs and cats with hyposomatotropism (both congenital and acquired) may also be in this range, which makes it difficult to document hyposecretion when relying solely on basal GH levels. Two dogs with congenital GH deficiencies did have consistently undetectable serum concentrations (Rijnberk et al, 1993). Currently, assessment of random basal GH concentrations is inadequate for documentation of hyposomatotropism; assessment of GH secretory capacity after stimulation of the pituitary somatotrophs is recommended to establish the diagnosis.
GH STIMULATION TESTS.
Several stimulation tests have been developed to evaluate the GH secretory capabilities of the pituitary. The most commonly used stimulation tests in the dog are the clonidine, xylazine, and GHRH stimulation tests and a combined pituitary anterior lobe function test using four releasing hormones: GHRH, thyrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), and corticotropin-releasing hormone (CRH) (Table 2-6). Use of these tests to assess GH secretion in cats has not yet been reported.
TABLE 2-6 GROWTH HORMONE STIMULATION TESTING PROTOCOLS
| Test | Description and Results |
|---|---|
| Xylazine Stimulation Test* | |
| Protocol | 100 μg/kg IV; plasma samples obtained before and at 15, 30, 45, and 60 minutes after administration of xylazine† |
| Normal results | Plasma GH, > 10 ng/ml 15 to 30 minutes after xylazine administration |
| Adverse reactions | Sedation (common), bradycardia, hypotension, collapse, shock, seizures |
| Clonidine Stimulation Test | |
| Protocol | 10 μg/kg IV; plasma samples obtained before and at 15, 30, 45, and 60 minutes after administration of clonidine† |
| Normal results | Plasma GH, > 10 ng/ml 15 to 30 minutes after clonidine administration |
| Adverse reactions | Sedation (common), bradycardia, hypotension, collapse, aggressive behavior |
| GHRH Stimulation Test | |
| Protocol | 1 μg/kg human GHRH IV; plasma samples before and at 10, 20, 30, 45, and 60 minutes after administration of GHRH |
| Normal results | Plasma GH, > 10 ng/ml 15 to 30 minutes after GHRH administration |
| Adverse reactions | None reported |
GH, Growth hormone; GHRH, growth hormone-releasing hormone.
* Currently preferred GH stimulation test.
† An abbreviated protocol in which plasma samples are obtained before and 20 and 30 minutes after stimulation can be done.
Interpretation of GH stimulation tests requires some knowledge of the conditions that may affect somatotroph responsiveness to various stimuli; this becomes especially important in the attempt to differentiate partial GH deficiency states from normal. A number of conditions may affect somatotroph responsiveness to stimuli in humans (Table 2-7) (Lazarus, 1984). Similar conditions have not been thoroughly evaluated in the dog, although hyperadrenocorticism, hypothyroidism, and some sex hormone imbalances may cause a reversible suppression of plasma GH concentrations (Peterson and Altszuler, 1981; Lothrop, 1988; Regnier and Garnier, 1995), and β-adrenergic antagonists potentiate the GH response to GHRH (Regnier et al, 1992).
TABLE 2-7 CONDITIONS THAT MAY AFFECT PITUITARY SOMATOTROPH RESPONSIVENESS TO GH STIMULATION TESTS IN HUMANS
| Decreased Responsiveness | Increased Responsiveness |
|---|---|
| Old age | Poorly regulated diabetes mellitus |
| Delayed puberty | Malnutrition |
| Obesity | Renal failure |
| Hypothyroidism | Liver cirrhosis |
| Thyrotoxicosis | Metastatic carcinoma |
| Hyperadrenocorticism | Estrogen therapy |
| Stress | |
| Glucocorticoids |
Modified from Lazarus L: In Donald RA(ed): Endocrine Disorders: AGuide to Diagnosis. New York, Marcel Dekker, Inc, 1984, p 273.
Clonidine Stimulation Test.
Clonidine is an α-adrenergic agonist that stimulates secretion of GHRH, which in turn stimulates secretion of GH. Injection of clonidine increases circulating GH and glucose concentrations and decreases plasma insulin concentrations in the dog (Hampshire and Altszuler, 1981). These effects are dose dependent. The use of clonidine to provoke GH secretion appears to be more reproducible and results in more dramatic changes in plasma GH concentrations than do the other stimulation tests. Currently, either the clonidine stimulation test or the GHRH stimulation test is preferred for diagnosing hyposomatotropism.
The dose of clonidine used in the test is somewhat variable and depends on the protocol established for the individual laboratory. An increase in GH may be seen at dosages as low as 3 μg/kg body weight given intravenously (Hampshire and Altszuler, 1981), although dosages of 10 μg/kg (Eigenmann, 1983), 16.5 μg/kg (Roth et al, 1980), and 30 μg/kg have been reported. The usual dosage is 10 μg/kg given intravenously. It should be kept in mind that larger dosages of clonidine (i.e., 16.5 and 30 μg/kg) produce a more pronounced and prolonged hyperglycemia but have a greater incidence of adverse reactions, including sedation, bradycardia, hypotension, collapse, and aggressive behavior (Lothrop, 1988; Eigenmann and Eigenmann, 1981a). Adverse reactions may last 15 to 60 minutes. If necessary, atropine can be used to reverse bradycardia, and the α-adrenergic antagonists phentolamine or yohimbine can be used to antagonize the hypotensive effects of clonidine (and xylazine). Plasma samples for GH determination should be obtained prior to clonidine administration and 15, 30, 45, 60, and 120 minutes after stimulation. GH is relatively stable for prolonged periods if frozen and maintained at −20° C.
In the normal dog, plasma GH concentrations should increase after clonidine administration, reaching peak concentrations 15 to 30 minutes after initiation of the test (Fig. 2-7) (Hampshire and Altszuler, 1981; Eigenmann, 1983). In healthy dogs, GH concentrations should exceed 10 ng/ml after clonidine stimulation. The GH concentration should then decline rapidly toward basal levels. Pituitary dwarfs and dogs with adult-onset complete GH deficiency do not respond to clonidine (see Fig. 2-7). In these dogs, no increase in plasma GH concentrations follows clonidine administration. A partial GH deficiency may be suspected whenever subnormal results are obtained (Table 2-8) (Eigenmann and Patterson, 1984).
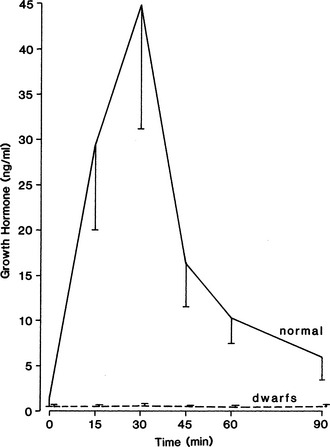

Evaluation of plasma glucose concentrations during clonidine-induced GH secretion may be helpful in evaluating somatotroph function. In the normal dog, plasma glucose concentrations increase after clonidine administration, an effect that is dose dependent (Hampshire and Altszuler, 1981). The peak increase in the plasma glucose concentration occurs approximately 90 minutes after clonidine administration. In contrast, no increase in plasma glucose occurs in hypophysectomized dogs or in dogs with hyposomatotropism (Eigenmann, 1981). The exact mechanism of the hyperglycemia is not known, although it seems likely to be a result of GH-induced insulin antagonism and decreased glucose uptake by cells.
Xylazine Stimulation Test.
Xylazine is a sedative analgesic that is structurally related to clonidine. Injection of xylazine increases circulating GH concentrations in normal dogs (Hampshire and Altszuler, 1981). The use of xylazine to provoke GH secretion is a suitable test for hyposomatotropism. Plasma samples are obtained prior to and 15, 30, 45, 60, and 120 minutes after intravenous administration of xylazine at a dosage of 100 to 300 μg/kg. Although the higher dose is typically recommended to ensure maximal stimulation of GH secretion, profound sedation, hypotension, shock, and seizures (rarely) may develop at these higher dosages. The lower dose (i.e., 100 μg/kg) is an effective GH stimulant but does not appear to increase the blood glucose concentration (Hampshire and Altszuler, 1981). The incidence of adverse reactions (e.g., sedation, bradycardia, hypotension, collapse) is also lower at this lesser dose. Interpretation of this test is similar to that of the clonidine stimulation test. In one study, the normal mean-peak serum GH concentration was 43.5 ± 40.8 ng/ml at 15 minutes after xylazine administration (Schmeitzel and Lothrop, 1990).
Growth Hormone-Releasing Hormone Administration Test.
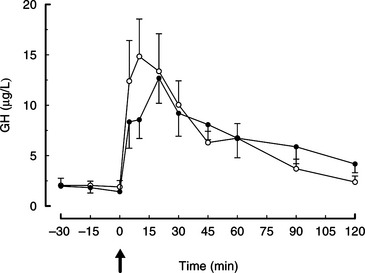
The use of GHRH has been evaluated as a diagnostic test for canine congenital and adult-onset GH deficiency (Lothrop, 1986; Rijnberk et al, 1993; Kooistra et al, 2000b). Administration of human GHRH (1 μg/kg IV; Peninsula Laboratories, San Carlos, CA) results in a rapid increase in plasma GH, with peak concentrations occurring 10 to 30 minutes after GHRH administration (Fig. 2-8; Abribat et al, 1989; Meij et al, 1996). The mean (± SE) plasma GH concentration was 14.7 ± 3.7 ng/ml at 10 minutes after GHRH administration (Meij et al, 1996). In eight German Shepherd dwarfs, basal GH concentrations were low and there was no increase in the plasma GH concentration after GHRH administration (see Fig. 2-4; Kooistra et al, 2000b). In six dogs with suspected adult-onset GH deficiency, basal GH concentrations were either low or within normal limits and there was no increase in GH after GHRH stimulation (Rijnberk et al, 1993). Adverse reactions were not observed after administration of GHRH. For the GHRH stimulation test, plasma samples for GH measurement should be obtained prior to and 5, 10, 20, 30, 45, and 60 minutes after intravenous administration of 1 μg of GHRH/kg body weight. Availability and cost are obvious disadvantages of GHRH.
Combined Anterior Pituitary Function Test.
A combined anterior pituitary function test can be used to assess pituitary function in dogs that are suspected of having pituitary dysfunction or that have undergone hypophysectomy for Cushing’s disease (see Chapter 6). The combined anterior pituitary function test consists of sequential, 30-second intravenous administrations of four hypothalamic releasing hormones in the following order and dosages: CRH, 1 μg/kg body weight; GHRH, 1 μg/kg; GnRH, 10 μg/kg; and TRH, 10 μg/kg (Meij et al, 1996). Plasma samples are collected prior to and 5, 10, 20, 30, 45, 60, 90, and 120 minutes after administration of the last releasing hormone for measurement of GH, ACTH, TSH, LH, FSH, and prolactin. In one study, the combined administration of these four hypothalamic releasing hormones caused no apparent inhibition or synergism with respect to the responses to GHRH, CRH, and TRH administered separately, but it did cause a 50% attenuation in the LH response compared with LH response to single GnRH administration (Meij et al, 1996). Adverse effects of the combined anterior pituitary function test include restlessness and nausea, which disappear shortly after administration of the releasing hormones. The combined anterior pituitary function test was used to document a combined deficiency of GH, TSH, and prolactin in 10 German Shepherd dwarfs (see Fig. 2-4; Hamann et al, 1999; Kooistra et al, 2000b). Availability and cost are obvious disadvantages of the combined anterior pituitary function test.
BASAL IGF-I CONCENTRATION.
IGF-I concentrations are low in dogs with pituitary dwarfism. In one study involving nine German Shepherd dwarfs, the mean plasma IGF-I concentration in normal adults was 280 ± 23 ng/ml; in immature normal animals, 345 ± 50 ng/ml; and in the dwarf dogs, 11 ± 2 ng/ml (Eigenmann et al, 1984c). Low plasma IGF-I concentrations have also been documented in three additional studies involving nine pituitary dwarfs (Rijnberk et al, 1993; Kooistra et al, 1998; Kooistra et al, 2000b). These findings support the theory of GH control over plasma IGF-I concentrations (see Fig. 2-2) and imply the important role that IGF-I plays in the regulation of growth. Radioimmunoassays for measurement of IGF-I in humans have been validated in dogs (Randolph et al, 1990) and cats (Church et al, 1994) and are commercially available. Measurement of serum IGF-I concentrations by radioimmunoassay provides a way to gain further evidence of GH deficiency as the cause of poor growth in dogs and cats when measurement of GH is not available. However, interpretation of results in dogs must take into consideration the size of the breed (i.e., smaller breeds have lower normal IGF-I concentrations and larger breeds have higher normal concentrations) (Eigenmann et al, 1984b).
THYROID FUNCTION.
Evaluation of thyroid gland function is imperative in any animal with suspected hyposomatotropism, because the clinical signs of hypothyroidism and hyposomatotropism are similar, making hypothyroidism an important differential diagnosis; also, TSH deficiency is the most common concurrent hormone deficiency in pituitary dwarfs with pituitary hypoplasia and combined hormonal deficiencies. In theory, the basal serum thyroxine, free thyroxine, and endogenous TSH concentrations should be low or undetectable in pituitary dwarfs with concurrent hypothyroidism. Unfortunately, an undetectable endogenous TSH concentration does not confirm pituitary TSH deficiency because the lower limit of the normal range using the TSH assay currently available extends below the sensitivity of the assay. In addition, low serum thyroxine or free thyroxine concentrations may be caused by extraneous factors affecting the thyroid gland function (e.g., euthyroid sick syndrome) rather than by pituitary TSH deficiency. The serum thyroxine concentration may also be within the reference range in a pituitary dwarf with impaired TSH secretion (Kooistra et al, 1998). For these reasons, evaluation of pituitary and thyroid gland function using thyrotropin-releasing hormone (TRH) stimulation testing is recommended (see Chapter 3, page 120). The results of TRH stimulation testing should be normal in dogs and cats with isolated GH deficiency. In dogs and cats with congenital hypothyroidism or pituitary dwarfism caused by combined anterior pituitary hormone deficiency, the baseline TSH, thyroxine, and free thyroxine concentrations should be low and there should be minimal to no increase in the concentrations of these hormones after administration of TRH (see Fig. 2-4). The reader is referred to Chapter 3 for more information on thyroid gland function tests.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


