Chapter 13 DIABETIC KETOACIDOSIS
Diabetic ketoacidosis (DKA) is a serious complication of diabetes mellitus. Before the availability of insulin in the 1920s, DKA was a uniformly fatal disorder. Even after the discovery of insulin, DKA continued to carry a grave prognosis, with a reported mortality rate in humans ranging from 10% to 30%. Despite expanding knowledge regarding the pathophysiology of DKA and the application of new treatment techniques for the complications of DKA, the mortality rate for this disorder remains at 5% to 10% in humans (Wagner et al, 1999; Masharani and Karam, 2001). DKA is a challenging disorder to treat, in part because of the deleterious impact of DKA on multiple organ systems and the common occurrence of concurrent, often serious, disorders that are primarily responsible for the high mortality rate of DKA. In humans, the incidence of DKA has not decreased, appropriate therapy remains controversial, and patients continue to succumb to this complication of diabetes mellitus. This chapter summarizes current concepts regarding the pathophysiology and management of DKA in dogs and cats.
PATHOGENESIS AND PATHOPHYSIOLOGY
GENERATION OF KETONE BODIES.
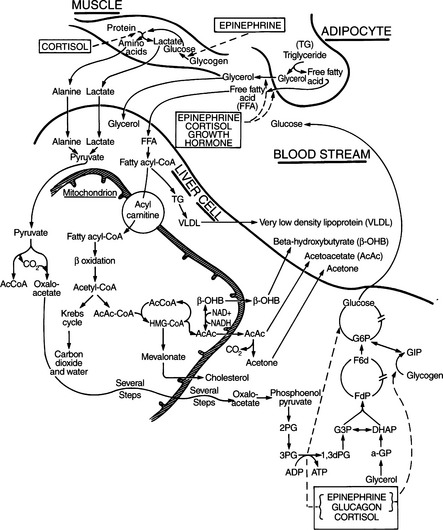
Ketone bodies are derived from oxidation of nonesterified or free fatty acids (FFA) by the liver and are used as an energy source by many tissues during periods of glucose deficiency. FFAs released from adipose tissue are assimilated by the liver at a rate dependent on their plasma concentration. Within the liver, FFAs can be incorporated into triglycerides, can be metabolized via the tricarboxylic acid (TCA) cycle to CO2 and water, or can be converted to ketone bodies (Fig. 13-1) (Hood and Tannen, 1994; Kitabchi et al, 2001). Oxidation of FFAs leads to the production of acetoacetate. In the presence of NADH, acetoacetate is reduced to β-hydroxybutyrate. Acetone is formed by spontaneous decarboxylation of acetoacetate (Fig. 13-1). These ketone bodies—acetoacetate, β-hydroxybutyrate, and acetone—are substrates for energy metabolism by most tissues in addition to fulfilling a biosynthetic role in the brain of the neonate (Zammit, 1994). The metabolism of ketone bodies is integrated with that of other substrates of energy metabolism, both in peripheral tissues and in the liver. However, excessive production of ketone bodies, as occurs in uncontrolled diabetes, results in their accumulation in the circulation and development of the ketosis and acidosis of ketoacidosis.
ROLE OF INSULIN DEFICIENCY.
The most important regulators of ketone body production are FFA availability and the ketogenic capacity of the liver (McGarry et al, 1989; Zammit, 1994). For the synthesis of ketone bodies to be enhanced, there must be two major alterations in intermediary metabolism: (1) enhanced mobilization of FFAs from triglycerides stored in adipose tissue and (2) a shift in hepatic metabolism from fat synthesis to fat oxidation and ketogenesis (Hood and Tannen, 1994; Kitabchi et al, 2001). Insulin is a powerful inhibitor of lipolysis and FFA oxidation (Groop et al, 1989). A relative or absolute deficiency of insulin “allows” lipolysis to increase, thus increasing the availability of FFAs to the liver and in turn promoting ketogenesis. Virtually all dogs and cats with DKA have a relative or absolute deficiency of insulin. In established diabetic animals in whom insulin is discontinued and in newly diagnosed diabetic animals who are diagnosed with ketoacidosis on initial examination, circulating insulin levels are low or undetectable. Some dogs and cats have serum insulin concentrations similar to those observed in normal, fasted nondiabetics (i.e., 5 to 20 μU/ml). However, such insulin concentrations are inappropriately low (“relative” insulin deficiency) for the severity of hyperglycemia encountered.
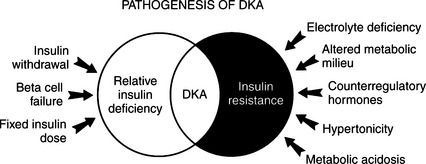
Some diabetic dogs and cats develop ketoacidosis despite receiving daily injections of insulin, and circulating insulin concentrations may even be increased. Insulin deficiency per se cannot be the sole physiologic cause for the development of DKA. In this group, a “relative” insulin deficiency is present. Presumably these dogs and cats have insulin resistance resulting from an increase in circulating glucose counterregulatory hormones (i.e., epinephrine, glucagon, cortisol, growth hormone) and an altered metabolic milieu (e.g., increased plasma FFAs and amino acids, metabolic acidosis). The ability to maintain normal glucose homeostasis represents a balance between the body’s sensitivity to insulin and the amount of insulin secreted by the beta cell or injected exogenously. With the development of insulin resistance, the need for insulin may exceed the daily injected insulin dose, and this leads to a predisposition for the development of DKA (Fig. 13-2).
ROLE OF GLUCOSE COUNTERREGULATORY HORMONES.
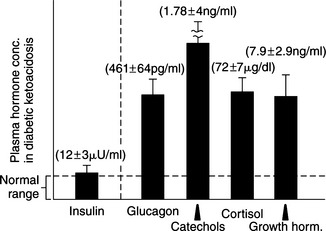
Circulating levels of epinephrine, norepinephrine, glucagon, cortisol, and growth hormone are typically markedly increased in humans with DKA, as are plasma FFA and amino acid concentrations (Fig. 13-3; Luzi et al, 1988). Increased circulating concentrations of these counterregulatory hormones cause insulin resistance, stimulate lipolysis and the generation of FFAs in the circulation, and shift hepatic metabolism of FFAs from fat synthesis to fat oxidation and ketogenesis (Fig. 13-3) (McGarry et al, 1989; Zammit, 1994). Glucagon is considered the most influential ketogenic hormone. Increased concentrations accompany ketotic states, and low concentrations blunt ketogenesis in ketogenic conditions (Hood and Tannen, 1994). Glucagon can directly influence hepatic ketogenesis. However, its effects still depend on substrate availability, and ketogenesis can occur in the absence of glucagon. Catecholamines are also important modulators of ketogenesis, primarily through stimulation of lipolysis. Both epinephrine and glucagon contribute to insulin resistance by inhibiting insulin-mediated glucose uptake in muscle and by stimulating hepatic glucose production through an augmentation of both glycogenolysis and gluconeogenesis (Cherrington et al, 1987; Cryer, 1993). Cortisol and growth hormone enhance lipolysis in the presence of a relative or absolute deficiency of insulin (see Fig. 13-1), block insulin action in peripheral tissues (Bratusch-Marrain, et al, 1982; Boyle, 1993), and potentiate the stimulating effect of epinephrine and glucagon on hepatic glucose output (Sherwin et al, 1980). An elevation in plasma FFA concentration and FFA oxidation inhibits insulin-mediated glucose uptake in muscle and stimulates hepatic gluconeogenesis (Thiebaud et al, 1982; Ferrannini et al, 1983). The combination of insulin deficiency and excesses in counterregulatory hormones also stimulates protein catabolism. Increased plasma amino acid concentrations impair insulin action in muscle and provide substrate to drive gluconeogenesis (Tessari et al, 1985). The net effect of these hormonal disturbances is accentuation of insulin deficiency through the development of insulin resistance, stimulation of lipolysis leading to ketogenesis, and stimulation of gluconeogenesis, which worsens hyperglycemia. All these factors lead to the eventual onset of clinical manifestations associated with DKA.
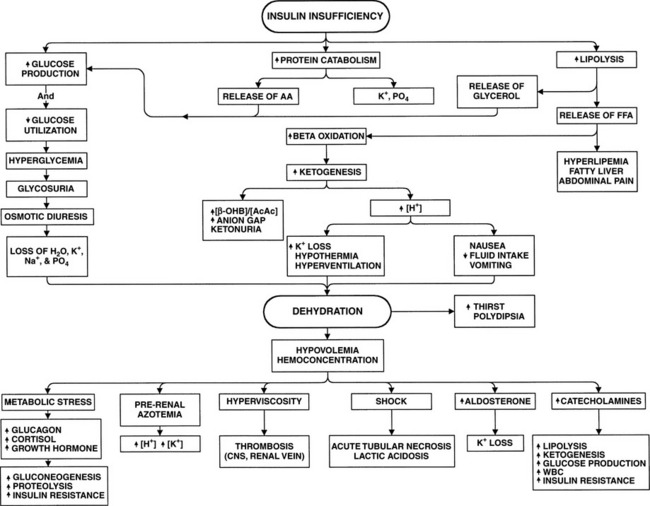
The body increases its production of the glucose counterregulatory hormones in response to a wide variety of diseases and stress situations. Although this response is usually beneficial, in DKA the activity of these hormones as insulin antagonists usually worsens hyperglycemia and ketonemia, provoking acidosis, fluid depletion, and hypotension. This condition progresses in a self-perpetuating spiral of metabolic decompensation (Fig. 13-4). It is rare for the dog or cat with DKA not to have some coexisting disorder, such as pancreatitis, infection, renal insufficiency, or concurrent hormonal disorder. These disorders have the potential for increasing glucose counterregulatory hormone secretion. For example, infection causes a marked increase in the secretion of cortisol and glucagon, heart failure and trauma result in increased circulating levels of glucagon and catecholamines, and fever induces secretion of glucagon, growth hormone, catecholamines, and cortisol (Kandel and Aberman, 1983; Feldman and Nelson, 1987). The recognition and treatment of disorders that coexist with DKA are critically important for successful management of DKA (see Fig. 13-2).
PHYSIOLOGIC CONSEQUENCES OF ENHANCED KETONE BODY PRODUCTION.
The physiologic derangements that accompany DKA are a direct result of relative or absolute insulin deficiency, hyperketonemia, and hyperglycemia (Table 13-1). In a short-term situation, the conversion of FFAs to ketone bodies is actually a positive metabolic development. Diabetes mellitus is interpreted physiologically as a state of starvation. With glucose deficiency, ketone bodies can be used as an energy source by many tissues. However, increasing plasma glucose and ketone concentrations eventually surpass the renal tubular threshold for complete reabsorption and spill into the urine, inducing an osmotic diuresis. During the development of DKA in humans, the daily urinary losses of glucose can be as much as 150 to 200 g, and ketone excretion rates average 20 to 30 g (Luzi et al, 1988; Ennis et al, 1994). Each gram of glucose excreted via the kidneys adds a solute load of approximately 6 mOsm. The lower molecular weight of ketones accounts for a greater osmotic load per gram, and excretion of ketones is responsible for one-third to one-half of the osmotic diuresis in humans with DKA (DeFronzo et al, 1994; Kitabchi et al, 2001). Moreover, the anionic charge on the ketones, even at a maximally acid urine pH, obligates the excretion of positively charged ions, such as sodium, potassium, calcium, and magnesium, to maintain electrical neutrality. The total urinary osmolar load from glucose and ketones alone can amount to 1500 to 2000 mOsm, an amount that is two- to three-fold greater than the normal daily urinary osmolar losses (DeFronzo et al, 1994). This increased solute excretion impairs the reabsorption of water throughout the proximal tubule and loop of Henle, lowering the concentration of sodium and chloride in the tubular lumen, causing an increased concentration gradient for their reabsorption, and thereby inhibiting their transport from lumen to blood. The result is an excessive loss of electrolytes and water, leading to volume contraction and underperfusion of tissues.

Insulin deficiency per se also contributes to the excessive renal losses of water and electrolytes. Physiologic increases in plasma insulin concentration augment salt and water reabsorption in both the proximal and distal portions of the nephron and enhance proximal tubular phosphate reabsorption (DeFronzo et al, 1994). Conversely, insulin deficiency leads to enhanced water and electrolyte excretion; it also leads to an accelerated rate of proteolysis, impaired protein synthesis, and negative nitrogen balance (DeFronzo and Ferrannini, 1992). The composition of the urine in a person undergoing an osmotic diuresis is similar to that of half-normal saline. Consequently, water losses typically exceed sodium losses. Significant losses of potassium and phosphorus also occur and may complicate therapy. Although increased urinary calcium and magnesium losses occur, they usually do not present a problem during management of DKA.
The formation of ketones by the liver is associated with the production of an equivalent number of hydrogen ions, which titrate the plasma bicarbonate concentration. As ketones continue to accumulate in the blood, the body’s buffering system becomes overwhelmed, causing an increase in arterial hydrogen ion concentration, a decrease in serum bicarbonate, and development of metabolic acidosis. Further loss of water and electrolytes occurs as a result of repeated bouts of vomiting or diarrhea combined with a lack of fluid intake, problems that often develop as the metabolic acidosis worsens (see Fig. 13-4). The excessive loss of electrolytes and water leads to further volume contraction, underperfusion of tissues, decline in the glomerular filtration rate (GFR), and worsening prerenal azotemia and dehydration. Hyperglycemic dogs and cats with reduced GFR lose the ability to excrete glucose and, to a lesser degree, hydrogen ions. Glucose and ketones then accumulate in the vascular space at a more rapid rate. The result is increasing hyperglycemia and ketonemia and worsening metabolic acidosis (see Fig. 13-4). The rise in the blood glucose concentration raises the plasma osmolality, and the resulting osmotic diuresis further aggravates the rise in plasma osmolality by causing water losses in excess of the salt loss. The increase in plasma osmolality causes water to be shifted out of cells, leading to cellular dehydration and the eventual development of obtundation and coma. The severe metabolic consequences of DKA, which include severe acidosis, hyperosmolality, obligatory osmotic diuresis, dehydration, and electrolyte derangements, ultimately become life-threatening.
SIGNALMENT
DKA is a serious complication of diabetes mellitus that occurs most commonly in dogs and cats with previously undiagnosed diabetes. Less commonly, DKA develops in an insulin-treated diabetic dog or cat that is receiving an inadequate dosage of insulin, often in conjunction with a concurrent infectious, inflammatory, or hormonal disorder. Because of the close association between DKA and newly diagnosed diabetes mellitus, the signalment for DKA in dogs and cats is similar to that for nonketotic diabetics (see Chapter 11, page 547 and Chapter 12, page 547). For the most part, DKA appears to be a disease of middle-aged and older dogs and cats, although DKA can be diagnosed at any age. In dogs DKA is diagnosed more commonly in females, whereas in cats it is more common in males. Any breed of dog or cat can develop DKA.
HISTORY AND PHYSICAL EXAMINATION
The history and findings on physical examination are variable, in part because of the progressive nature of the disorder and the variable time between DKA onset and owner recognition of a problem. The spectrum ranges from ketonuric dogs and cats that are otherwise healthy, are eating, and have not yet developed metabolic acidosis (i.e., diabetic ketosis [DK]) to ketonuric dogs and cats that have developed severe metabolic acidosis (i.e., DKA), have severe signs of illness, and are moribund.
A complete and careful physical examination is critically important in any ketoacidotic animal (see Fig. 13-2). The initial physical examination should focus on an evaluation of the status of hydration, on the extent of central nervous system (CNS) depression, and on a careful search for any initiating cause for diabetic decompensation and ultimate ketoacidosis. Diabetic dogs and cats frequently suffer from concurrent infections, pyometra, pancreatitis, cholangiohepatitis, renal insufficiency, cardiac disease, or other insulin-antagonistic disorders (Bruskiewicz et al, 1997). A careful history, physical examination, and judicious use of laboratory tests can, in most circumstances, identify underlying concurrent disorders, lead to appropriate treatment, and increase the likelihood of successful therapy.
Common physical findings include dehydration, depression, weakness, tachypnea, vomiting, and sometimes a strong odor of acetone on the breath. With severe metabolic acidosis, slow, deep breathing (i.e., Kussmaul respiration) may be observed. Gastrointestinal signs of vomiting, abdominal pain, and distention may be identified and must be differentiated from similar signs associated with pancreatitis, peritonitis, or other intra-abdominal disorders. The vomiting and abdominal pain that accompany DKA are usually acute in onset, beginning after diabetes mellitus is well established. Conversely, a history of intermittent abdominal pain or vomiting occurring over a period of days or weeks before examination should raise suspicion for a separate abdominal problem, especially chronic pancreatitis. Additional physical examination findings associated with uncomplicated diabetes mellitus (e.g., cataracts, peripheral neuropathy) may also be identified in the dog or cat with DKA (see Chapter 11, page 492, and Chapter 12, page 547).
ESTABLISHING THE DIAGNOSIS OF DIABETIC KETOSIS AND KETOACIDOSIS
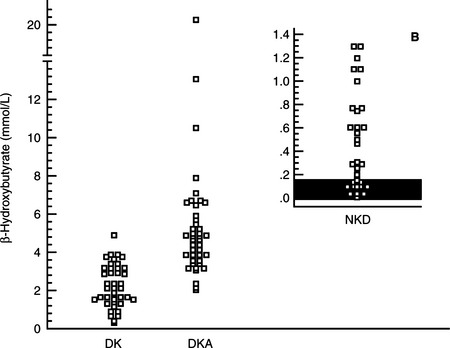
A diagnosis of diabetes mellitus requires the presence of appropriate clinical signs (i.e., polyuria, polydipsia, polyphagia, weight loss) and documentation of persistent fasting hyperglycemia and glycosuria. Measurement of the blood glucose concentration using a portable blood glucose monitoring device (see page 512) and testing for the presence of glycosuria using urine reagent test strips (e.g., KetoDiastix; Ames Division, Miles Laboratories Inc., Elkhart, IN) allows the rapid confirmation of diabetes mellitus. The concurrent documentation of ketonuria establishes a diagnosis of diabetic ketosis or ketoacidosis. The subsequent documentation of metabolic acidosis differentiates DKA from DK. Urine reagent test strips for ketonuria measure acetoacetate and acetone. If ketonuria is not present but DK or DKA is suspected, blood can be tested for the presence of acetoacetate and acetone with Acetest tablets (Ames Division, Miles Laboratories, Inc, Elkhart, IN) or the presence of β-hydroxybutyrate using a bench-top chemistry analyzer. The nitroprusside reagents used for the rapid identification of ketoacidemia and ketoaciduria (Acetest and Ketostix) detect only ketone (–C=O) groups (i.e., acetoacetate, acetone). In humans, the predominate ketone body produced during DKA is β-hydroxybutyrate. The β-hydroxybutyrate:acetoacetate ratio can range from 3:1 to 20:1 depending on the severity of hypovolemia, tissue hypoxia, and lactic acidosis (Li et al, 1980; Goldstein, 1995). In the presence of circulatory collapse, an increase in lactic acid can shift the redox state to increase β-hydroxybutyrate at the expense of the readily detectable acetoacetate. In theory, severe hyperketonemia could be underestimated or even undetected if urine reagent test strips or blood tests using the nitroprusside reagent are used to identify DK or DKA. In one study, differences in the serum concentration of β-hydroxybutyrate were identified between dogs with nonketotic diabetes mellitus, DK and DKA, suggesting that measurement of serum β-hydroxybutyrate could be of value in diagnosing and monitoring ketosis and ketoacidosis in diabetic dogs (Fig. 13-5; Duarte et al, 2002).
The aggressiveness of the diagnostic evaluation and treatment of a dog or cat with DKA is dictated primarily by results of the history and physical examination. A diagnosis of severe DKA is indicated in dogs and cats with systemic signs of illness (i.e., lethargy, anorexia, vomiting), a physical examination revealing dehydration, depression, weakness, and/or Kussmaul respiration; blood glucose concentration greater than 500 mg/dl; or severe metabolic acidosis as diagnosed by a total venous CO2 or arterial bicarbonate concentration less than 12 mEq/L. History and physical examination findings are subjective, however, and a veterinarian may not be able to obtain quick acid-base information. Therefore a diagnosis of severe DKA is often initially based on having an ill dog or cat with glucose and ketones in the urine. Further diagnostic tests are necessary to care for these animals properly, but therapy can and should proceed while remaining tests are pending.
IN-HOSPITAL DIAGNOSTIC EVALUATION
OVERVIEW.
The laboratory evaluation of “healthy” ketotic diabetic dogs and cats is similar to that for nonketotic diabetics (see Chapter 11, page 493 and Chapter 12, page 548). The healthy ketotic diabetic can usually be managed conservatively, as contrasted with the needs of an extremely ill DKA pet. To aid in the formulation of a treatment protocol, the clinician must perform a group of critically important studies in the ill ketoacidotic diabetic. Without these evaluations, an animal’s metabolic status cannot be adequately assessed and the clinician cannot have sufficient knowledge of the specific abnormalities that require correction. The minimum required tests include urinalysis, hematocrit, total plasma protein concentration, blood glucose, venous total carbon dioxide or arterial acid-base evaluation, blood urea nitrogen (BUN) or serum creatinine, and serum electrolytes (Na, K, Ca, PO4).
These studies are needed for the immediate assessment and intensive care of the animal. Knowing the results of these tests allows for proper choice of fluid therapy as well as for corrections that must be made with respect to electrolyte alterations, acidosis, and renal function. Other data, such as diagnostic imaging, electrocardiogram, or additional laboratory studies, may be needed for a complete medical assessment. However, the “ketoacidosis profile” provides the information necessary to begin proper emergency therapy.
Urinalysis and urine culture
The urinalysis can serve several purposes simultaneously. The most obvious reason for obtaining a urine sample is to identify glycosuria and ketonuria. A diagnosis of severe DKA should be made in the dehydrated, ill dog or cat with glycosuria and ketonuria. Fluid therapy should be initiated (see page 597) while confirmatory blood glucose and acid-base studies and determination of serum electrolyte concentrations are performed.
The minimum data base (ketoacidosis profile)
BLOOD GLUCOSE.
Although the average blood glucose concentration in dogs and cats with DKA is approximately 500 mg/dl, values can range from close to 200 mg/dl to concentrations that are characteristic of hyperosmolar coma (i.e., >1000 mg/dl; Fig. 13-6). Because hepatic production of glucose is excessive in dogs and cats with DKA and relative or absolute deficiencies of insulin always exist, it is likely that the degree of hyperglycemia is determined primarily by the severity of dehydration and corresponding decrease in GFR (Foster and McGarry, 1983). Evidence suggests that blood glucose concentrations become extremely elevated (i.e., >500 mg/dl) only when the extracellular fluid (ECF) volume has decreased to the point that urine flow and the capacity to excrete glucose are impaired. Studies have shown a marked reduction in the blood glucose concentration when humans with DKA were treated solely with fluids (i.e., no insulin; Owen et al, 1981), findings also seen clinically in diabetic dogs and cats (Fig. 13-7). An association has been made in humans with diabetes between the maximum attainable blood glucose concentration and the severity of reduction in GFR (Table 13-2; Kandel and Aberman, 1983).
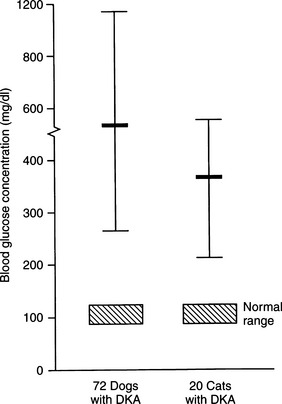
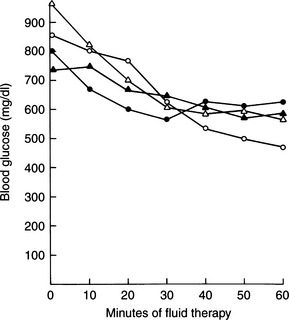
TABLE 13-2 MAXIMUM SERUM GLUCOSE CONCENTRATIONS IN DIABETIC HUMANS FOR GIVEN GLOMERULAR FILTRATION RATES
| Glomerular Filtration Rate (ml/min) | Serum Glucose Concentration (mg/dl) |
|---|---|
| 108 | 400 |
| 72 | 600 |
| 54 | 800 |
| 36 | 1200 |
| 27 | 1600 |
From Kandel G, Aberman A: Selected developments in the understanding of diabetic ketoacidosis. Can Med Assoc J 128:392, 1983.
ACID-BASE STATUS.
Metabolic acidosis is one of the hallmark clinical pathologic changes in DKA and is the direct result of an excess accumulation of ketone bodies in the blood. Excessive serum ketones can overwhelm the body’s buffering system, causing an increase in arterial hydrogen ion concentration, a decrease in serum bicarbonate, and a progressively worsening metabolic acidosis (Fig. 13-8).
Recognition of acidosis is usually straightforward with the use of arterial blood gas or venous total CO2 determinations (Table 13-3). In DKA, the severity of changes in arterial blood gas or venous total CO2 depends on the duration and severity of hyperketonemia at the time of presentation to the veterinarian. Arterial pH can range from 7.2 to as low as 6.6. Dogs and cats with arterial blood pH values less than 7.0 have life-threatening DKA and are difficult to treat successfully. A tremendous amount of controversy surrounds therapy directed specifically at the acidosis component of DKA. A discussion on the pros and cons of bicarbonate therapy for animals with severe DKA is found on page 602.
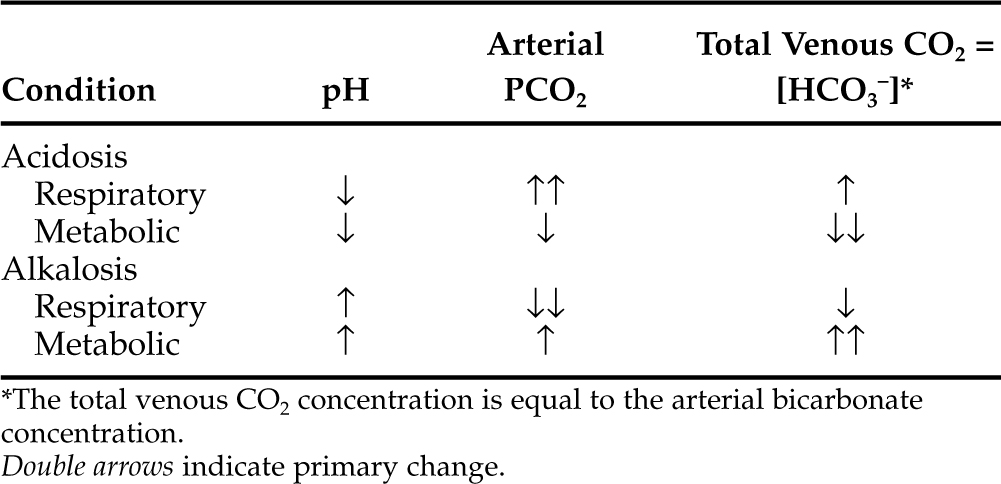
SERUM SODIUM CONCENTRATION.
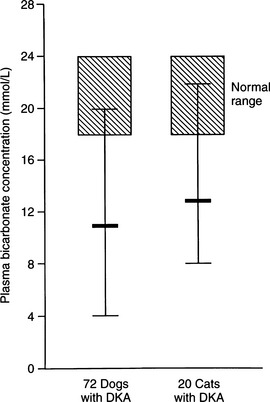
The serum sodium concentration is a reflection of the relative amounts of water and sodium present in the body. With rare exception, dogs and cats with DKA have significant deficits in total body sodium, regardless of the measured serum concentration. In 72 dogs with DKA, 62% were hyponatremic and only 7% were hypernatremic (Fig. 13-9). Similarly, in 42 cats with DKA, 80% were hyponatremic and only 5% were hypernatremic (Bruskiewicz et al, 1997).
Hyponatremia results from excessive urinary sodium loss caused by the osmotic diuresis induced by glycosuria and ketonuria. Because insulin enhances renal sodium reabsorption in the distal portion of the nephron, its absence results in sodium wasting. Hyperglucagonemia, vomiting, and diarrhea also contribute to the sodium loss in DKA (Foster and McGarry, 1983). Severe hypertriglyceridemia may occasionally cause factitious hyponatremia. Severe hypertriglyceridemia can be recognized by the presence of lipemia retinalis on ophthalmoscopic examination or by the presence of gross lipemia on visual inspection of serum or plasma. Saline and Ringer’s solutions have adequate sodium quantities for replacement of severe sodium deficiencies and are recommended as the initial fluids of choice for the treatment of severe DKA (see page 597).
It is important to consider the severity of hyperglycemia when assessing the severity of hyponatremia in the dog or cat with DKA. Because glucose penetrates cells poorly in the absence of insulin, an increase in ECF glucose concentration creates a transcellular osmotic gradient that results in the movement of water out of the cells and a corresponding reduction in the plasma sodium concentration. In general, for each 100 mg/dl increment in serum glucose above the normal range, the plasma sodium concentration decreases by approximately 1.6 mEq/L (DiBartola, 2000). Conversely, as insulin therapy drives glucose into the cells, water will follow and the plasma sodium concentration will increase.
SERUM POTASSIUM CONCENTRATION.
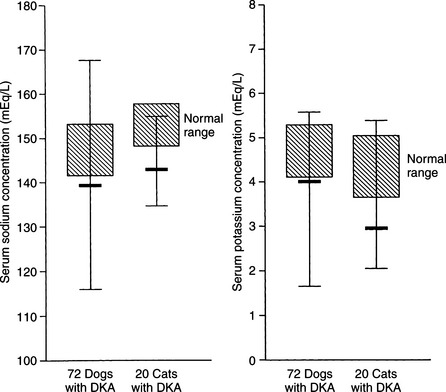
During DKA, intracellular dehydration occurs as hyperglycemia and water loss lead to increased plasma and ECF tonicity and a shift of water out of cells. This shift of water is also associated with a shift of potassium into the extracellular space (Fig. 13-10; Kitabchi et al, 2001). Potassium shifts are further enhanced by the presence of acidosis and the breakdown of intracellular protein secondary to insulin deficiency. Entry of potassium into cells is also impaired in the presence of insulinopenia. The osmotic diuresis induced by glycosuria and ketonuria causes marked urinary losses of potassium. Secondary hyperaldosteronism induced by plasma volume contraction, gastrointestinal losses, and decreased dietary intake augment the potassium deficiency (Kitabchi et al, 2001). As a consequence, most dogs and cats with DKA have a net deficit of total body potassium. Serum potassium concentrations can be decreased, normal, or increased, depending on the duration of illness, renal function, and previous nutritional state of the dog or cat. Most dogs and cats with DKA have either normal or decreased serum potassium concentrations on pretreatment testing (see Fig. 13-9). In 72 dogs and 42 cats with DKA, 43% and 67% were hypokalemic and 10% and 7% were hyperkalemic, respectively, at the time DKA was diagnosed (Bruskiewicz et al, 1997).
Knowing the serum potassium concentration and status of renal function is critical when deciding on the aggressiveness of potassium supplementation in the IV fluids. Polyuric DKA animals are predisposed to severe hypokalemia, and oliguric/anuric animals are predisposed to severe hyperkalemia. Insulin treatment causes a marked translocation of potassium from the ECF to the intracellular fluid (ICF) compartment, which when combined with continuing renal and gastrointestinal loss, can cause severe hypokalemia during the initial 24 to 48 hours of treatment. DKA dogs and cats that are hypokalemic on initial evaluation require aggressive potassium supplementation to their intravenous fluids to replace deficits and to prevent worsening hypokalemia (see page 600).
BLOOD UREA NITROGEN AND CREATININE.
The BUN and serum creatinine concentrations are commonly elevated in DKA (Fig. 13-11) and are useful indicators of the severity of volume depletion. When evaluated in conjunction with the urine specific gravity and serum calcium:phosphorus ratio, they can also help to identify concurrent primary renal failure versus prerenal azotemia. In addition, the initial BUN or creatinine concentration can serve as a measure of the success of fluid therapy. A rapidly falling BUN is consistent with proper fluid therapy, good urine output, and a dog or cat that has prerenal azotemia. The increased BUN that is slowly declining or static suggests inadequate fluid therapy or primary renal failure.
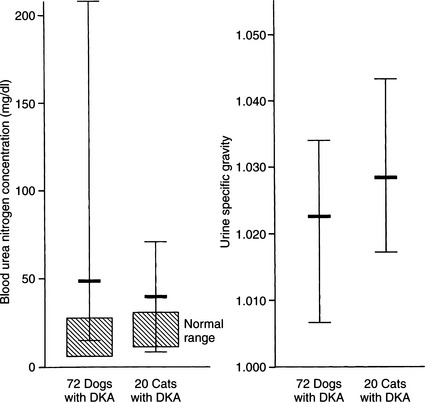
Increased serum creatinine concentrations may be spurious, resulting from interference by acetoacetate (one of the ketone bodies). At serum acetoacetate concentrations of 8 to 10 mmol/L, the serum creatinine concentration is falsely elevated 3 to 4 mg/dl by the Beckman autoanalyzer method and 1 to 2 mg/dl by the Technicon autoanalyzer method. If the serum creatinine concentration is determined manually, elevated serum ketone levels do not interfere (Molitch et al, 1980; Nanji and Campbell, 1981).
SERUM OSMOLALITY.
Hyperosmolality is a potentially serious development in DKA, one that can have profound effects on CNS function and consciousness. Of all the factors related to stupor or altered consciousness, including the serum levels of glucose, ketones, or arterial pH, the serum osmolality correlates best with the level of consciousness in humans with DKA. “Clouded consciousness” is an extremely subjective finding in humans, making recognition of such a problem guesswork in dogs or cats. Nevertheless, veterinarians and owners can usually recognize “depression” in the dog and cat, and in our experience, the severity of this sign roughly correlates with the severity of hyperosmolality.
Fortunately, severe hyperosmolality (>350 to 400 mOsm/kg) occurs infrequently in dogs and cats with DKA, in part because of the concurrent prevalence of hyponatremia (see Fig. 13-9). Serum sodium concentration and, to a lesser extent, potassium and glucose are the primary determinants of effective serum osmolality. Hyperosmolality, if present, usually resolves with intravenous isotonic fluid and insulin therapy, although correction of the hyperosmolar state must be done slowly to minimize the shift of water from the extracellular to the intracellular compartment.
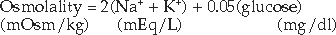
ANION GAP.
The metabolic acidosis stemming from hyperketonemia is an anion gap acidosis, which must be differentiated from other causes of anion gap acidosis (e.g., lactic acidosis, renal failure, ethylene glycol intoxication) and from hyperchloremic acidosis (Narins et al, 1994). The anion gap is calculated by subtracting the negatively charged anions, chloride and bicarbonate, from the most important positively charged cations, sodium and potassium. The normal anion gap for dogs and cats is 12 to 16 mEq/L (Feldman and Rosenberg, 1981). Anything greater than 16 mEq/L indicates the presence of an anion gap acidosis. The unmeasured anions that comprise the normal anion gap include albumin and other circulating proteins, sulfate, phosphate, and a variety of organic acids.
The typical diabetic dog or cat in ketoacidosis has an anion gap that ranges from 20 to 35 mEq/L, and the increment in the anion gap above baseline approximates the decrement in serum bicarbonate concentration (Adrogué et al, 1982). A number of factors can disrupt the normal stoichiometry between acid retention, increment in the anion gap, and decrease in serum bicarbonate concentration (Table 13-4). Such disruption is common in DKA because urinary loss of ketoanions causes a disproportionately greater decrease in serum bicarbonate concentration compared with the increment in anion gap. In these cases, chloride replaces the missing ketoanion, and a component of hyperchloremic acidosis commonly accompanies the anion gap acidosis (Table 13-5; Adrogué et al, 1982; 1984). Dogs and cats that are volume-contracted tend to have a pure anion gap acidosis because the decrease in GFR and tubular avidity for sodium reabsorption limits the urinary loss of ketone bodies. Conversely, animals with DKA able to maintain salt and water intake avoid severe volume depletion. These animals have variable degrees of hyperchloremic acidosis because of the urinary excretion of ketone salts and a concomitant retention of chloride (Table 13-5).
TABLE 13-4 INFLUENCES OF COMMON METABOLIC DISORDERS ON THE CALCULATED ANION GAP*
* These disorders are frequently associated with metabolic acidosis.

Completing the data base
COMPLETE BLOOD COUNT.
The complete blood count (CBC) in uncomplicated DKA usually reveals a leukocytosis without evidence of toxic neutrophils (Bruskiewicz et al, 1997). The leukocytosis may occur secondary to the release of “stress” hormones or severe inflammation, especially if an underlying pancreatitis is present. White blood cell counts greater than 30,000/μl, the presence of toxic or degenerative neutrophils, or a significant left shift toward immaturity of the cells supports the presence of a severe inflammatory and/or infectious process as the cause of the leukocytosis. The red blood cell count and hematocrit should be consistent with hemoconcentration secondary to dehydration. A hematocrit below 35% in these typically volume-contracted animals should arouse suspicion that blood loss has occurred or that significant bone marrow suppression or another problem has resulted in anemia.
LIVER ENZYMES AND TOTAL BILIRUBIN.
Clinical pathologic abnormalities associated with the liver are common in dogs and cats with DKA and are usually caused by hepatic lipidosis, pancreatitis, severe acidosis, hypovolemia, hypoxia, sepsis, and, less commonly, extrahepatic biliary obstruction caused by acute severe pancreatitis, chronic hepatitis, and cholangiohepatitis (see page 608). Aspartate transaminase (AST), alanine transaminase (ALT), and serum alkaline phosphatase (SAP) concentrations are usually increased (see page 493). Less commonly, fasting serum bile acid concentrations and serum total bilirubin concentrations may be increased, although icterus is not common. In some dogs and cats, a specific cause for the abnormal liver enzyme patterns cannot be identified. No significant correlation exists between the serum level of hepatic enzymes and the presence or severity of the hepatopathy.
PANCREATIC ENZYMES.
Because acute and chronic pancreatitis is so common in dogs and cats with DKA, a diagnostic evaluation for its existence is always warranted (Bruskiewicz et al, 1997). In our experience, abdominal ultrasound is the single best diagnostic test for identifying pancreatitis in the dog or cat with DKA (see Fig. 12-10, page 549). Measurement of serum lipase concentration and trypsin-like immunoreactivity (TLI) are also commonly recommended. In theory, dogs and cats with active inflammation of the pancreas should have an increase in serum lipase and TLI. Unfortunately, serum lipase concentrations and serum TLI do not always correlate accurately with the presence or absence of pancreatitis (Hess et al, 1998; Swift et al, 2000; Gerhardt et al, 2001). Pancreatic enzyme concentrations can be increased in dogs and cats with a histologically confirmed normal pancreas and normal in dogs and cats with histologically confirmed inflammation of the pancreas, especially when the inflammatory process is chronic and mild. For these reasons, interpretation of serum lipase and TLI results should always be done in context with the history, physical examination findings, and additional findings on the laboratory tests. Recognition of concurrent pancreatitis in the dog or cat with DKA has important implications regarding initial fluid therapy, subsequent dietary therapy, and prognosis. Fortunately, aggressive fluid therapy is the cornerstone of treatment for both DKA and pancreatitis. In most cases, treating one disease also treats the other.
CALCIUM AND PHOSPHORUS.
The serum calcium and phosphorus concentrations are usually normal in the diabetic dog or cat with “uncomplicated” DKA. If concurrent primary renal failure is present, the serum calcium concentration is typically normal, whereas the serum phosphorus concentration is increased. In pancreatitis, mild hypocalcemia may occur secondary to an acute shift of calcium into soft tissues such as muscle as a result of altered membrane integrity, alterations in thyrocalcitonin secretion or tissue responsiveness to parathyroid hormone, hypomagnesemia, hypoproteinemia, and deposition of calcium in plaques of saponified fat. Hypercalcemia supports the existence of concurrent disease, including chronic renal failure (see Chapter 16).
Attention has been directed to serum phosphorus concentrations in animals with DKA, especially during the initial 24 hours of treatment. Phosphate, along with potassium, shifts from the intracellular to the extracellular compartment in response to hyperglycemia and hyperosmolality (see Fig. 13-10; Kitabchi et al, 2001). Osmotic diuresis subsequently leads to enhanced urinary phosphate losses. Serum phosphorus concentrations can be decreased, normal, or increased, depending on the duration of illness and renal function. Most dogs and cats with DKA have either normal or decreased serum phosphorus concentrations on pretreatment testing. Hypophosphatemia (<3.0 mg/dl) was identified at initial presentation in 24% of 72 dogs and 48% of 42 cats with DKA (Bruskiewicz et al, 1997). In contrast, hyperphosphatemia (>6.0 mg/dl) was identified in 14% and 26% of DKA dogs and cats, respectively, and usually occurred in conjunction with renal failure. Insulin treatment causes a marked translocation of phosphorus from the ECF to the ICF compartment. Within 24 hours of initiating treatment for DKA, serum phosphorus concentration can decline to severe levels (i.e., <1 mg/dl) as a result of the dilutional effects of fluid therapy, the intracellular shift of phosphorus following the initiation of insulin therapy, and continuing renal and gastrointestinal loss (Willard et al, 1987). Clinical signs may develop when the serum phosphorus concentration is less than 1.5 mg/dl, although signs are quite variable and severe hypophosphatemia is clinically silent in many animals. Hypophosphatemia primarily affects the hematologic and neuromuscular systems in the dog and cat (Forrester and Moreland, 1989). Hemolytic anemia is the most common and serious sequela to hypophosphatemia. Hypophosphatemia may decrease erythrocyte concentration of ATP and/or alter red blood cell membrane lipids, which increases erythrocyte fragility, leading to hemolysis (Shilo et al, 1985; Adams et al, 1993). Hemolysis is usually not identified until the serum phosphorus concentration is 1 mg/dl or less. Hemolytic anemia can be life-threatening if not recognized and treated. Neuromuscular signs include weakness, ataxia, and seizures, as well as anorexia and vomiting secondary to intestinal ileus. Phosphate therapy is indicated if clinical signs of hemolysis are identified or if the serum phosphorus concentration is less than 1.5 mg/dl (see page 601).
MAGNESIUM.
The osmotic diuresis of DKA may cause significant urinary losses of magnesium and the development of hypomagnesemia (serum total magnesium concentration <1.75 mg/dl; serum ionized magnesium concentration measured by ion-selective electrode <1.0 mg/dl) (Norris et al, 1999). In addition, the nature of the translocation of magnesium between the ICF and ECF compartments is similar to potassium in that factors which promote a shift of potassium into the ICF compartment (e.g., alkalosis, insulin, glucose infusion) promote a similar shift in magnesium. During therapy for DKA, the serum total and ionized magnesium concentration can decline to severely low levels (i.e., less than 1 mg/dl and 0.5 mg/dl, respectively) as a result of the dilutional effects of fluid therapy and the intracellular shift of magnesium after the initiation of insulin therapy (Norris et al, 1999). Clinical signs of hypomagnesemia do not usually occur until the serum total magnesium concentration is less than 1.0 mg/dl, and even at these low levels, many animals remain asymptomatic. A magnesium deficiency can result in several nonspecific clinical signs, including lethargy, anorexia, muscle weakness (including dysphagia and dyspnea), muscle fasciculations, seizures, ataxia, and coma (Martin et al, 1993; Abbott and Rude, 1993; Dhupa and Proulx, 1998). Concurrent hypokalemia, hyponatremia, and hypocalcemia occur in animals with hypomagnesemia, although the prevalence of these electrolyte abnormalities may differ between species. These electrolyte abnormalities may also contribute to the development of clinical signs. Magnesium is a cofactor for all enzyme reactions that involve ATP, most notably the sodium-potassium ATPase pump. Deficiencies in magnesium can lead to potassium wastage from the body and the resultant hypokalemia may be refractory to appropriate potassium replacement therapy. Magnesium deficiency inhibits PTH secretion from the parathyroid gland, resulting in hypocalcemia (Bush et al, 2001). Magnesium deficiency causes the resting membrane potential of myocardial cells to be decreased and leads to increased Purkinje fiber excitability, with the consequent generation of arrhythmias (Abbott and Rude, 1993). ECG changes include a prolonged PR interval, widened QRS complex, depressed ST segment, and peaked T waves. Cardiac arrhythmias associated with magnesium deficiency include atrial fibrillation, supraventricular tachycardia, ventricular tachycardia, and ventricular fibrillation. Hypomagnesemia also predisposes animals to digitalis-induced arrhythmias.
Unfortunately, assessing an animal’s magnesium status is problematic because there is no simple, rapid, and accurate laboratory test to gauge the total body magnesium status. Approximately 1% of total body magnesium is present in serum; as a result, serum magnesium concentrations do not always reflect the total body magnesium status. A normal serum magnesium concentration may exist despite an intracellular magnesium deficiency. However, a low serum magnesium concentration does support the presence of a total body magnesium deficiency. Magnesium exists in three distinct forms in serum: an ionized fraction, an anion-complexed fraction, and a protein-bound fraction. A serum ionized magnesium concentration determined by using an ion-selective electrode is more accurate in assessing total body magnesium content and is recommended over measurement of serum total magnesium (Norris et al, 1999b). Fortunately, hypomagnesemia is not usually a clinically recognizable problem during management of DKA and magnesium supplementation is not recommended unless hypomagnesemia is documented in dogs and cats with complications that have been associated with hypomagnesemia (e.g., persistent lethargy and anorexia; refractory hypokalemia).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


