, Monika Hassel2 and Maura Grealy3
(1)
Centre of Organismal Studies, University of Heidelberg, Heidelberg, Germany
(2)
Spezielle Zoologie, Universität Marburg FB Biologie, Marburg, Germany
(3)
Pharmacology and Therapeutics, National University of Ireland Galway, Galway, Ireland
Signals controlling development eventually turn on genes – or they turn genes off. In this chapter we are dealing with genes directing global events, and with genes paving the path to differentiation and executing the differentiation. This is an issue so extensive and diverse that any textbook can only provide a basic introduction.
Box 12.1: On the Nomenclature Regarding Genes and Their Products
We first briefly recapitulate some rules for correct notation outlined in the appendix of Chap. 2.
The designation of genes, and of the mRNA derived from them, is written in italics. In the history of Drosophila genetics capital letters were used to denominate dominant genes (e.g. Antennapedia), while recessive genes were written with small letters (e.g. bicoid) provided statements about the relative expressivity of genes were possible. In addition, it has become common usage, but not imperative rule, to contract the designation of genes to a few – most frequently three – letters (bicoid to bcd).
Guidelines for human gene nomenclature were proposed by Wain et al. (2002).
The proteins derived from these genes are written in standard letters, increasingly frequently in capital letters (e.g. BICOID), as it is done in this textbook. This allows, for instance, to distinguish right away the protein WINGLESS, as coded from the wingless allele, from a wingless fly.
Shorthand symbols indicating the genetic constitution of an organism used in genetics are plus (+) for functional wild type alleles, minus (−) for defect alleles ( loss-of-function alleles). Using these symbols in diploid organisms the following constitutions are possible: +/+ homozygous for the wild-type allele; −/− homozygous for the defect allele, and +/− heterozygous combining one wild-type with one defect allele. For instance the designation bc −/+ or bcd /+ means, that a defect bcd allele and a functional wild-type bcd allele are present. The specimens with the constitution −/− are frequently called k.o. (knock out) mutants. Dominant mutations may generate gain-of-function alleles.
Please note: The name of genes is usually derived from the phenotype (appearance) of mutants. Therefore, the designation can be very misleading if one would deduce the function of the wild type allele from the mutant-derived name. The wild-type gene dorsal of Drosophila, for instance, and the wild-type DORSAL protein coded by the dorsal gene are not required for the development of dorsal but of ventral structures. In the (dorsal-/-) mutant no functional protein is produced and dorsal instead of ventral structures are made, for whatever reasons.
Many names of genes refer to the phenotype without giving hints to the particular function of the protein. For example mutations in the Antennapedia complex may result in flies bearing legs instead of antennae. The function of the ANTENNAPEDIA protein, however, was mysterious for a long time, and eventually only revealed by arduous research work.
Box 12.2: Genetic and Molecular Methods in Developmental Biology
Detecting Genes Relevant for Development
We speak of relevant genes if their products essentially contribute to the control of development but also of genes the expression of which denotes a developmental path and the products of which are useful markers of differentiation paths leading to distinct cell types. In case of muscle tissue, for example, the members of the myoD/myogenin family are regulatory genes, while the appearance of muscle-specific actin and myosin filaments indicates terminal differentiation in progress.
Genes relevant to development can be identified by two broad approaches: forward genetics and reverse genetics. Forward genetics traces from the phenotype to the genotype. The mutant phenotype may be produced as a result of natural variation or breeding, or as a result of mutagenesis. The mutants are then analysed by positional cloning to find the gene responsible for the phenotype. Following this the wild-type gene is searched for, including its control region (enhancer, promoter).
Reverse genetics proceeds in the opposite direction to forward genetics; we start with a known sequence and trace the phenotype. Due to genome sequencing, we now have sequences for many genes whose function we do not know. In reverse genetics the function of a specific known gene is manipulated, for example by gene silencing, knockout or overexpression, and the resulting phenotype examined to find the function of the gene.
How Mutants Are Generated and Analysed
Profound analysis of many mutants is only realisable with reasonable expenditure if the animals propagate sexually, have a short life-cycle duration (preferentially less than 4 months), can be kept in large numbers without demanding much space and physical and human resources. Also the ability to make “knockouts” of selected genes has become a critically important technique. The procedures have been most frequently and successfully applied in the fly Drosophila and the small nematode Caenorhabditis elegans which provides the convenient possibility to store defined genetic stocks in the freezer. The zebrafish Danio rerio and the medaka fish Orycias latipes are easily crossed, but need expensive aquarium facilities for large-scale genetics. With considerably higher expenditures genetics is done with mice, proxy of humans. The technician time and space required for profound genetic analyses, removal of embryos from the maternal animal and thorough search for altered traits indicating a developmentally relevant mutation are very high.
Chemical Mutagenesis. This is the classical procedure. As a rule, mutagenic agents are administered to male individuals only. The agents are mixed with the food or given into the drinking water; fishes have to swim in solutions of such agents. Progeny is carefully examined for abnormal phenotypes, and not only the F1-generation. Since in all likelihood a mutation affects only one allele of a pair of homologous alleles, as a rule mutations come to light only in the F2 when the mutated allele is present in some individuals in homozygous condition (Fig. 12.1). Today, targeted genetic fingerprinting can detect mutations already in heterozygous F1, and facilitates selection of individuals for continuing cross-breeding.
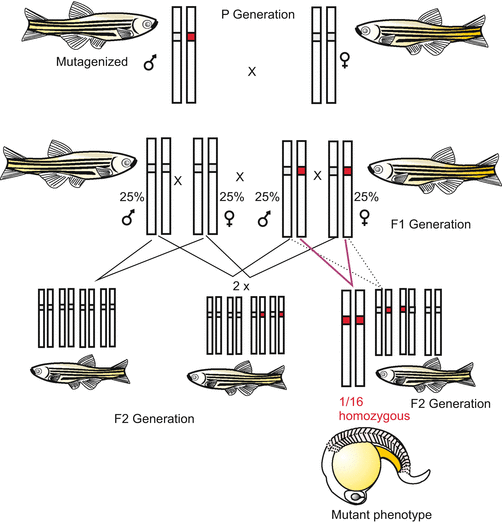
Fig. 12.1
Cross-breeding to arrive at homozygosity of an induced, recessive mutation. Recessive mutations become phenotypically obvious in the homozygous condition only. Statistically this condition is found in 1/16 of the individuals of the F2 generation
The particular skill a researcher needs to develop is a sense for judging which mutation is of specific significance. A defect-causing mutation in a general household gene will evoke considerable, often lethal disturbances, but these are not development-specific. Indicative for relevance are, for example, localized defects or cessation of development at a certain stage. A strong indication can be deduced from the genetics. If the Mendelian rule of reciprocity is infringed and the genetic constitution of the mother decisive (but not of the father or the child itself) strong interest is justified. Then, classic cross-breeding must aid in narrowing down the chromosomal locus affected by the mutation.
From Mutations to Genes. Searching for the actually affected gene is tedious, time-consuming, and its definite identification difficult. With regard to successful strategies for searching and the large array of available genetic tools we must refer to textbooks of molecular genetics. Suggested key words are: linkage group, chromosomal walk, positional cloning; correlation of a trait with AFLP (Amplified Fragment Length Polymorphism).
The gene searched for is definitively identified only if a functional analysis succeeds.
The gene is expressed at the right time and the correct place.
The defect is curable through injection of wild-type mRNA or injection of protein produced (in the lab) using the wild-type gene (rescue experiment).
Anti-sense RNA directed against the wild-type gene or RNA interference (RNAi, see Fig. 12.6) simulates the mutant phenotype (phenocopy) in wild-type embryos.
Gene products (mRNA or the protein derived from it) generate additional structures if injected at wrong (ectopic) locations, or they alter developmental fates of body regions dramatically, for example, by inducing head formation at the posterior body pole (see Drosophila, Fig. 4.32).
P-element Mutagenesis and Transgenesis. In Drosophila melanogaster (and similarly in Caenorhabditis elegans) a different kind of mutagenesis facilitates search for the mutated gene. This procedure is known as P-element transformation. A P-element is a virus-like transposon that is naturally present in wild-type Drosophila melanogaster. The P transposon is used to create genetically modified flies. P-elements occur in two forms: (1) in form of ring-like plasmids or (2) linearly inserted in the host’s genome. The transposon is flanked by terminal inverted repeats which are important for its insertion and excision. Insertion takes place by a “cut and paste” mechanism, and can occur at many sites in the genome causing disruption of genes. For movement of the element a transposase is required and a transposase gene is naturally part of the P-element. Complete transposons with integrated transposase are life-threatening for a Drosophila population, because in the permanent presence of transposase the P-elements can move beyond the fly’s control and destroy genes (Many natural populations died out, but some survived because they developed some kind of inhibition to uncontrolled movements or had transposons lacking the transposase gene).
For mutagenesis in the lab two types of plasmids were isolated from Drosophila strains and engineered according to the researchers desire: plasmid type 1 lacking the transposase gene, and plasmid type 2 comprising transposase but lacking the transposon-type flanking sequences and therefore unsuitable to be inserted into chromosomes. Based on plasmid type 1 constructs were made containing, between the flanking sequences, a Drosophila reporter gene, often a red-eye marker (the product of the white gene) and, optionally, any other transgene chosen by the researcher, for example a selectable marker gene, often some kind of antibiotic resistance. Both types of plasmids, the plasmid type 1 lacking transposase and the plasmid type 2 containing transposase, are injected at the posterior pole of freshly laid eggs before pole cells form (Fig. 12.2). A pole cell (primordial germ cell) that accidentally takes up a transposon type 2 is qualified to produce transposase enzyme. If the cells in addition has taken up a transposon type 1 this transposon will be inserted into the genome, mediated by the transposase, and often disrupt a protein-coding gene. Such fly strains can be used to generate mutated offspring only for a limited time; they will not survive in the long term if the transposase is not outcrossed. (Transposase-bearing individuals can cause genetic instability of a strain). Once the gene of interest, for instance a transgene, has been inserted and the transposase gene removed, the transgene-containing plasmid is no longer mobile and the exchanged mutated gene and/or the introduced transgene is a permanent part of the genetic heritage of the respective strain.
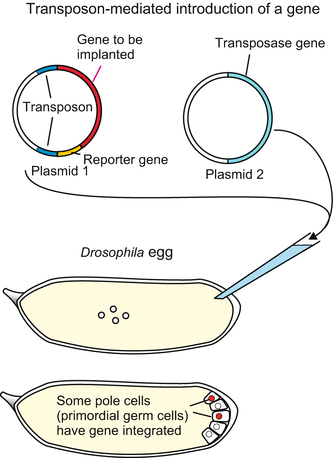
Fig. 12.2
P-element-induced mutagenesis in Drosophila. (a) When injected into the cytoplasm of a still uncleaved egg, P-element-type plasmids can be taken up by the forming pole cells, can integrate into chromosomes aided by transposase, insert into genes and cause mutations by disrupting the message. Offspring that inherited a plasmid can be recognized if the plasmid brings along a marker gene, for example the bacterial LacZ gene which codes for β-galactosidase, an enzyme that can produce a blue dye in an established lab protocol
Following mutagenesis and selection of mutants of interest from progeny DNA is extracted and cut into pieces by means of restriction enzymes to establish a genomic library (see following section). This is screened for sequences containing sequences that are partially indicative of the fly, partially indicative for the inserted plasmid, containing a fly-specific sequence linked with a part of the transposon-type flanking sequence or a part of the reporter gene. Such sequences, containing both host-specific and plasmid-specific sequences, could only derive from those genes into which the transposon was inserted. These partial sequences of the affected host gene can, after their cloning and labelling, be used as probes to search for overlapping sequences in the genomic library. With some diligence and luck a whole set of overlapping sequences can be collected until the whole gene including its promoter and enhancers is complete. Available databases of the complete genome facilitates the search considerably.
How Genomic or EST Databanks Help to Quickly Identify and Isolate a Gene or its Relatives
Genomic Banks. To establish a genomic bank the entire DNA of an organism is extracted, fragmented in pieces by a collection of restriction enzymes, or randomly by applying shearing forces, splitting long DNA helices into small workable portions. The fragments are ligated into a vector, typically a bacteriophage. Vectors have multiple restriction enzyme consensus sites to either side of the insert so that the inserted DNA fragment can later be cut out. Using these phage vectors the DNA fragments are transferred into bacteria and ‘cloned’ (=multiplied, amplified) by breeding the bacteria. Samples of the bacteria are stored frozen: a ‘genomic library’ or ‘genomic bank’ is established.
Sequencing and Databases. For sequencing, the fragments are cut out from the bacterial clones by restriction enzymes, and subjected to any of the available (automatized) sequencing procedure.
To establish a complete library and database, many thousands of fragments must be sequenced. Bioinformatics provide tools to reconstruct from this chaotic collection of sequences the entire genome by assembling overlapping sequences.
More and more methods are available or in development to quickly establish unknown DNA sequences, that is to determine the precise order of nucleotides within a stretch of DNA, with high speed using fluorescence-based sequencing methods and automatic machines. Selected key words for the reader’s own enquiries : Second and third generation sequencing or high-throughput sequencing, Single-molecule real-time sequencing, Sequencing by ligation (SOLiD), Cyclodextrin-hemolysin nanopore technology, Nanoball sequencing, Sequencing with mass spectrometry, RNA polymerase-based sequencing (RNAP), RNA sequencing (RNA seq); Microscopy-based techniques.
Using appropriate methods the search for putative genes is simplified and speeded up, in particular the search for genes with high sequence similarity such as members of gene families or other homologous genes, but also detecting novel genes is facilitated.
However since large genomes including the human genome contain giant sections of non-protein-coding sequences, effort and expenditure are large, but the yield is low, if one is looking for a distinct protein-coding gene (without the help of an attached marker as in case of P element transformation). By contrast, under aspects of gene regulation non-protein-coding sequences are of particular interest.
The feasibility of large-scale genome sequencing has led to an increasing number sequenced entire genomes. Yet, genome sequencing as such does not reveal much about the function of a gene. To unravel the function of a particular gene one has to mutate it, or to manipulate it in some other way, as discussed below. Important hints as to the function can be deduced from EST banks.
EST Projects and cDNA Libraries. EST is the abbreviated term of Expressed Sequence Tag. In EST projects the sum of all expressed genes, the transcriptome, of an entire organism, of a developmental stage, or of an organ or a tissue, is analysed. The starting material is extracted mRNA; the mRNA is transcribed into cDNA using an enzyme called reverse transcriptase, and often subjected to a first amplification step by PCR (Polymerase Chain Reaction). The resulting cDNA molecules are engineered so that they have restriction enzyme recognition sites at each end of the molecule, which allows them to be inserted into a vector and to be excised after cloning in bacteria. By means of vectors the cDNA specimens are transferred into bacteria and cloned (amplified) to establish a cDNA bank (library). In such libraries only sequences are present derived from mRNA and, thus, mRNA that was with high probability transcribed from genes relevant for the developmental stage and biological source under investigation. Supplemented by parallel in-situ hybridisation studies EST projects enable the survey of gene expression patterns in the course of development and cell differentiation.
How Reverse Genetics Allow Tracing Genes
There are, as mentioned above, only a few organisms which allow the identification of a large array of developmentally-relevant genes by mutagenesis and subsequent crossings, and from which many genes have been cloned and sequenced.
Of course, researchers working with organisms less or not suited for genetics, for example Xenopus laevis or Hydra, seek possibilities to trace such genes and to study their expression also in their organism of choice. Such possibilities are provided by reverse genetics. If in the genome of the organism of choice, let’s say in Xenopus, a gene is present displaying sequence similarity to a known gene of, for example, Drosophila (100–70 % sequence identity), reverse genetics helps. One chooses a cloned piece of the Drosophila gene, for example the conserved homeobox of the Antennapedia gene, flags it by coupling an easily identifiable marker, and uses it as a heterologous (derived from another species) probe to screen a genomic or cDNA bank (library) made from DNA or mRNA extracted from Xenopus in a search for similar, autologous sequences. Today, this is done on a DNA chip (see below). If such a sequence is found it is amplified by molecular cloning or by means of Polymerase Chain Reaction PCR-based amplification, and sequenced.
However, reverse genetics cannot hunt out completely unknown sequences such as those endemic to the species in question. Unknown genes of relevance can be tracked down with the following procedure.
From mRNA Via cDNA to Unknown, Novel Genes and Study of Their Expression
Search for Differentially Expressed Genes by Means of cDNA Subtraction
A standard technique to identify and isolate differentially expressed genes is cDNA subtraction that enables enrichment of cell- or stage-specifically expressed mRNA. First mRNA is extracted from two different cell populations (e.g. early versus late gastrula). During synthesis of cDNA using reverse transcriptase the arising cDNA derived from a reference source (e.g. late gastrula) can be labelled with biotin. If one now adds a small amount of cDNA to be studied (e.g. derived from the early gastrula) and conditions are chosen (heating) to transiently separate DNA into single strands, all cDNA molecules present in both sources will find a partner and hybridise, when conditions (cooling) allow, forming double strands. Since they are marked with biotin they bind to streptavidin-coated beads (streptavidin binds biotin) and can be removed. As only unlabelled cDNA, specific for the source of interest (here early gastrula), is left unbound and survives, one speaks of cDNA subtraction. The remaining, specific cDNA molecules are cloned into a vector and eventually characterized using the tools of molecular biology.
Using the subtraction technique, however, all gene expression that takes place in overlapping stages or simultaneously in the two compared sources remains hidden; in our example mRNA present in early as well as in late gastrula, gets lost. Several configuration levels have been proposed promising higher efficiency, for example differential display (DDRT-PCR. However, these methods have largely been replaced by chip-based methods including DNA-microarrays and more recently by RNA-Seq combined with software to identify differentially expressed genes (for example DESeq).
DNA-Microarrays (DNA-Chips)
DNA microarrays, also known as DNA chips, are applied to comparative genome analysis, diagnosis of genetically-caused diseases and studies of differential gene expression. Further, DNA microarrays can be used, for example, to measure changes in gene expression levels, to detect single nucleotide polymorphisms (SNPs), or to genotype organisms in biodiversity studies.
In the sense of molecular biology a microarray is a collection of microscopic DNA spots firmly attached to a solid surface in a well-defined pattern on a grid (spotted microarrays, Figs. 12.3 and 12.4). Each DNA spot contains picomoles (10−12 moles) of a specific DNA sequence, known as a probe or reporter or ‘oligo’. The spotted and immobilized sequences are characteristic single-stranded sections of genes or other DNA elements complementary to ‘target’ sequences to be detected and identified. In standard microarrays, the probes are synthesized and then attached via surface engineering to a solid surface by a covalent bond to a chemical matrix which covers a glass or a silicon chip. Alternatively, microarrays can be constructed by the direct synthesis of oligonucleotide probes on solid surfaces. Each DNA probe is plotted twice.
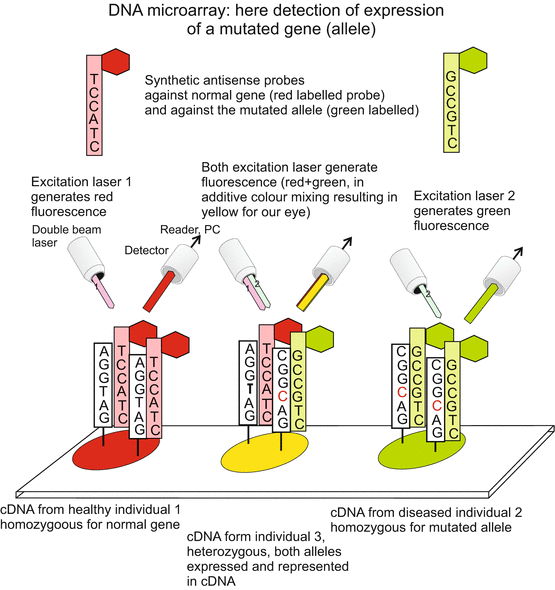
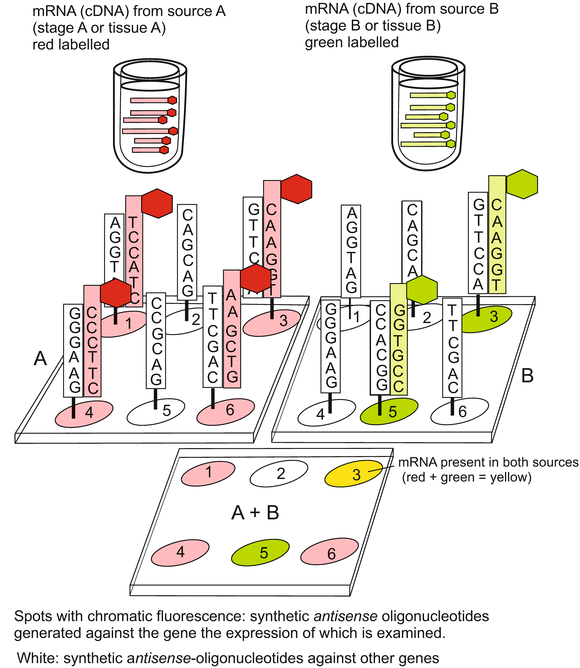
Fig. 12.3
DNA microarray (DNA chip) I. On a support, typically on a small silicon plate (chip), small amounts (spots) of DNA are loaded as reference probes, in the present case single-strand cDNA corresponding to selected genes and/or mutated alleles of these genes. The reference cDNA probes are fixed on the spots. In the second step single-stranded cDNA samples from sources to be assayed are labelled with fluorescing dyes and hybridized with the reference cDNA. In the example shown it is checked whether the examined sources contain mutated alleles or sequences identical to the reference probes. From the colour of the reflected laser beam it can be deduced whether a mutated allele is present, and whether it is present in the assayed source in homozygous or heterozygous condition
Fig. 12.4
DNA microarray (DNA chip) II. Here sequences of DNA are fixed onto the support spots that are unique sequences characteristic for the selected genes. One single chip may be loaded with thousands of spots representing thousands of different genes. Messenger RNA extracted from various sources to be compared is transcribed in cDNA and thereby labelled with different fluorescing dyes. In the example shown cDNA from the first source is marked red, cDNA from the second source labelled green. Through hybridisation it can be examined whether and to what extent the two sources contain identical or different mRNAs
The target sequences to be analysed attach to the probes on the chip by hybridisation, and if they match perfectly they remain attached even under high-stringency conditions. Since an array can contain tens of thousands of probes, a microarray experiment can accomplish many genetic tests in parallel. The target nucleic acids are usually linked to fluorescent dyes. Normally there are two samples which are compared, one tagged with red fluorescent dye and the other with green. The probe-target hybridisation pairs are detected and quantified by measuring the wavelength ratios, typically the red-to-green fluorescence ratio, and strength of fluorescence emitted by the spots.
Which probes are spotted onto the chips, and how many, depends on the question to be answered, on exploratory work previously done, and on previous achievements. Probes could be, for example
original DNA sequences amplified by polymerase chain reaction PCR,
synthetic DNA sequences (oligonucleotide microarrays) or
cDNA samples of differentially expressed genes obtained by cDNA subtraction.
If one wants, for example, to know whether in a suspicious-looking tissue mRNA of an oncogene (cancer gene) is present, then the following procedure can be used: Probes of synthetic oligonucleotides are prepared, each probe being characteristic for a distinct oncogene, and immobilized on pre-defined positions of the grid. Then mRNA is extracted from the suspicious tissue, transcribed into cDNA and linked with a fluorescence dye. The thus marked collection of cDNA is hybridized with the oligo spots on the array. Because you know which spot on the grid represents which oncogene, you can deduce from the pattern of fluorescing spots which oncogenes had been active in the tissue sample. Furthermore, because each probe was plotted twice on the array, digitalized comparisons assessing the signal strengths allows evaluating which oncogene was expressed particularly strongly.
Possible applications of microarrays are manifold. Any conceivable application can be realized, whereby always sense-antisense partners must be chosen for hybridisation and, as a rule, mRNA is used in the form of its cDNA:
DNA versus DNA from a sample to be diagnosed versus reference DNA; by choosing appropriate reference DNAs it can be explored whether a mutated allele is present in the sample and, if so, whether it is present in homozygous or heterozygous condition.
DNA versus DNA from two different individuals or species to see whether they share genes with high sequence similarity (putative homologue genes).
DNA versus mRNA (transcribed into cDNA) from different sources. The DNA probes represent a collection of genes the expression of which is investigated, for example, as outlined above, a collection of oncogenes; mRNA is extracted from putative cancer tissue and normal tissue, transcribed into cDNA and labelled. By analysing the pattern of signals it can be inferred which gene had been active in the samples. (This case is illustrated in Fig. 12.3.)
DNA versus mRNA (transcribed into cDNA) from different developmental stages. The DNA probes represent developmentally relevant genes the expression of which is investigated; mRNA is extracted from different developmental stages, let’s say from a gastrula and from a blastula. By comparing the strength of the hybridisation signals hints can be deduced as to which gene activities are down-regulated or up-regulated in the course of a developmental process (This case is illustrated in Fig. 12.4).
mRNA versus mRNA from different sources, for instance as exploratory work for cDNA subtraction but also to identify genes up- or down-regulated in diseased vs normal states, in drug-treated vs untreated cells or animals.
However, microarrays do not provide information on where exactly in the embryo a gene is expressed. For that mRNA would have to be extracted from single, exactly located cells. A very useful, supplementing technique to localize expression sites is the in-situ hybridisation technique described in the following section.
Screening by Whole Mount In-Situ Hybridisation Visualizes Cell Type and Stage-Specific Gene Expression in Whole Animals
A strategy aiming at a complete expression analysis has been called in situ screening or expression screening, and proved successful in Xenopus. From the tissue of interest, let’s say the upper blastopore lip of the early gastrula, mRNA is isolated and used to construct a plasmid-cDNA bank in which the orientation of the inserted and cloned genes within the plasmids is identical and known. The used plasmid vector possesses an inserted promoter at its 3′-terminus that binds a T3 RNA polymerase, and a further promoter at its 5′-terminus where a T7 RNA polymerase can dock. These promoters enable the in-vitro synthesis of labelled (for example digoxygenin- or biotin-labelled) sense- and antisense-RNA. (Antisense-RNA runs antiparallel to mRNA and by hybridisation both together can form a double strand). The labelled antisense-RNA is used as a probe in the procedure known as in-situ hybridisation, to demonstrate endogenous mRNAs that are expressed at various locations in the embryo, and at different times (Fig. 12.5).
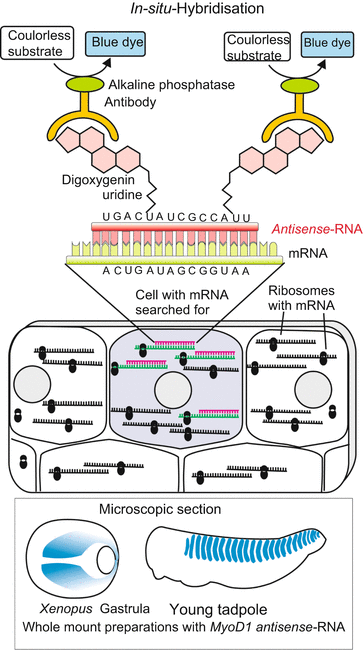
Fig. 12.5
In-situ-hybridisation. The technique is used to visualize the presence of a certain endogenous mRNA in (chemically) fixed samples of cells or whole objects (for example whole embryos or microscopic tissue slices). In the lab synthetic antisense-RNA is generated linked with—in the present case—digoxigenin-UTP. The antisense-RNA binds (hybridizes) to the sense-RNA (=mRNA), if present in the object. The hybrids are detected by means of anti-digoxigenin antibodies. These are coupled to an enzyme that catalyses the formation of a blue or fluorescing dye
Whenever possible in-situ hybridisation is performed in intact embryos or animals (whole mount preparations), so that the three-dimensional arrangement (pattern of expression) of the labelled cells is preserved. If the specimen is too large or not transparent it is dissected to prepare microscopic slices. From a series of two-dimensional pictures the three-dimensional arrangement of the structures must subsequently be reconstructed with the aid of a computer using an image processing program.
Messenger RNAs, that are differentially expressed (region-specific, stage-specific or cell type-specific), can be investigated further. Compared to the method of cDNA subtraction this strategy has the advantage that also mRNA from genes which are simultaneously expressed in several cell populations, territories and developmental stages is rendered visible and does not remain hidden. The targeted search for groups of genes that are expressed in identical or similar spatial and temporal patterns (synexpression groups) and therefore are likely functionally linked has provided instructive results.
There are more means and procedures to study gene expression, among them a “shining example” in which particular reporter genes are involved. An example will be outlined below in the section “Demonstration of proteins without using genetic engineering”.
Protein Microarrays and Immuno-Fluorescence
Microarrays and in-situ hybridisation refer to the level of DNA and RNA. Yet, since transcription and translation can considerably diverge in space and time, and proteins often do not remain at the place of their synthesis, the detection and localization of proteins is also required. For that purpose also several types of micro-arrays can be made. Partners for mutual binding can be antigen-antibody, enzyme-substrate, receptor-ligand, or protein-protein in general.
Proteins extracted from a biological source are immobilized on spots of the chip and solutions of defined partner proteins such as a fluorescently labelled antibody, is added. Or conversely, a collection of defined test proteins, for example a collection of various selected antibodies, is fixed on the solid surface in an ordered arrangement, and protein sample extracted from the biological source in question added. One of the two binding partners, the antibodies for example, are coupled with fluorescent dyes, and the pattern of fluorescent spots, indicating binding, is analysed by means of a fluorescence reader.
The counterpart of detecting mRNA in microscopic slices by in-situ hybridisation is detecting the location of defined proteins in microscopic preparations by immuno-cytochemistry using antibodies (see Fig. 12.11).
Studies of Gene Function, Applications
How the Function of Genes Can Be Tested Without Conducting Genetics
Inhibition Through Antisense-Oligonucleotides. Translation of selected mRNA species in living cells can be inhibited through hybridisation (annealing) with short, complementary single-stranded DNA pieces (antisense-oligonucleotides) if they pair with sequences at, or close to, translation codons (ATG). The protein is no longer synthesized. Lack of the respective protein can cause a phenocopy of knock-out mutations, that is, evoke symptoms including traits in the appearance identical with those seen in mutants.
Antisense RNAs can be of natural origin. Natural antisense transcripts (NATs) are a group of RNAs encoded within a cell that have transcript complementarity to other RNA transcripts. They are often found among the category of the small miRNAs or siRNAs. Antisense species complementary to mRNA can suppress translation of the corresponding mRNAs or induce their degradation (Fig. 12.6); they are known to be transcribed for that purpose in development and cell differentiation.
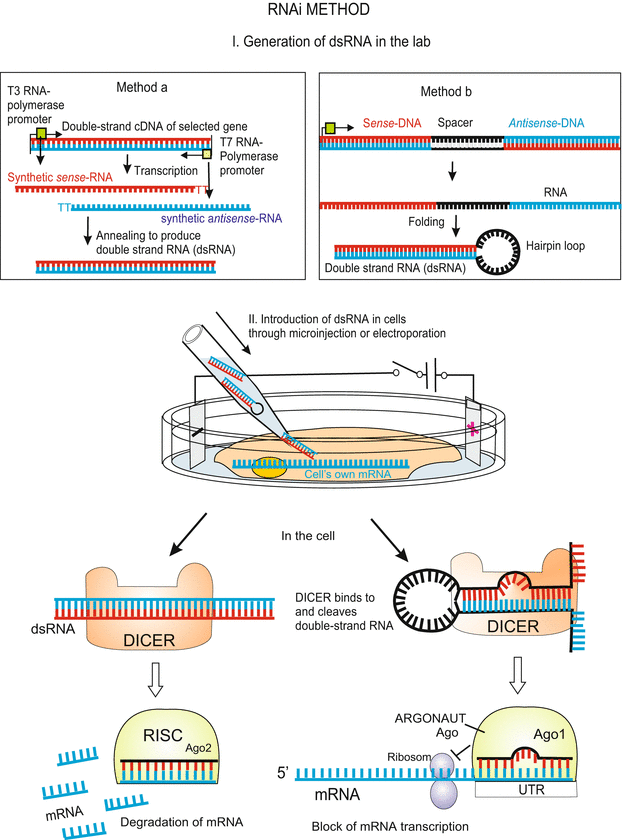
Fig. 12.6
RNAi method to deactivate a gene function on the level of its mRNA. In the laboratory a corresponding double-stranded RNA (dsRNA, with or without a hairpin loop) is synthesized and introduced into the target cell – as a rule an egg cell or a stem cell. In the cell the DICER complex cuts out small RNA pieces from the dsRNA. Transferred to an ARGONAUT protein Ago (see Fig. 12.21) these pieces can bind to complementary sequences in the untranslated starting region UTR of the targeted mRNA and activate there a RISC complex. In cooperation with a set of additional proteins (not shown) the mRNA is cleaved into small pieces which are not translated. A dsRNA with a hairpin loop which does not fully fit to the UTR of the mRNA leads to a block of its translation. The procedures make use of natural ways to stop translation. For details of these procedures see Fig. 12.21
For introduction in cells, synthetic, single-stranded antisense–oligonucleotides are enclosed in lipid micelles suited to merge with cell membranes and to release their content into the cells (lipofection = liposome transfection). Alternatively, the oligos are transferred from concentrated solutions into the host cells by making them transiently permeable by a pulse-type application of an externally applied electrical field (electroporation or electropermeabilisation). This procedure is highly efficient also for the introduction of foreign genes in tissue culture cells, especially mammalian cells. In studies on gene function with antisense oligonucleotides usually not natural oligos are used because these are quickly degraded; instead artificial, chemically modified nucleotides (phosphorothioate, morpholinos) are linked together yielding a class of rather stable antisense probes. These are more resistant to degradation by nucleases than are the natural transcripts. Morpholinos are widely used in developmental biology to knock down genes. They can be targeted against the ATG start site to block translation, or against a splice site to block mRNA splicing. They are injected into the embryo at the one-cell stage and persist for about 3–4 days. This allows time to see the effects of transient gene knockdown in models with rapid development, for example zebrafish.
Inhibition by RNA Interference (RNAi) Using siRNA. RNA interference (RNAi) is a method to induce destruction of specific mRNA molecules. RNAi enables the knockdown of gene function without targeted mutagenesis (Fig. 12.6). Historically, it turned out that the method, developed in the laboratory, imitates natural phenomena, especially the function of natural siRNA (see Fig. 12.21). The method gained currency when in 2006, A. Fire and C. C. Mello shared the Nobel Prize for their work on RNA interference in the nematode C. elegans, and protocols were developed to apply the method in other organisms as well.
In practise, a sequence derived from a gene (or a cDNA) is used as template to synthesize corresponding, complementary sense and antisense RNA strands, both folded together to form double-stranded dsRNA molecules. Introduced into a cell or organism by microinjection or lipofection, dsRNA is recognized as exogenous genetic material and activates the RNAi pathway. An internal complex of enzymes known as DICER fragments the dsRNA into short pieces, each comprising 21–28 nucleotides, generally called siRNA (defined as short inhibiting RNAs or small interfering RNAs). Subsequently, these pieces are transferred to proteins of the ARGONAUTE (Ago) family by an endogenous mechanism. One of the two strands of the siRNA attaches to the UTR (UnTranslated Region) in the start region of an existing mRNA. If the siRNA and mRNA strands match, the Ago protein is complemented by further proteins and a complex called RISC (RNA-Induced Silencing Complex) is established. The RISC complex functions as a nuclease and the mRNA is cleaved resulting 21- to 25-mers. These are materials from which new and more siRNA pieces are made. The process proceeds avalanche-like, or in scientific terms, autocatalytically. The primary reaction gives rise to further siRNAs which attack further parts of the mRNA yielding the next set of siRNAs, and so forth, until all endogenous mRNA is destroyed. No more protein can be made. The phenotype corresponds to the phenotype of a knock out mutation, like the phenotype evoked by antisense nucleotides, except that a DNA mutation is heritable. Modifications of the method allow, instead of down-regulation of gene functions, activation of translation beyond the normal level, a kind of gene overexpression.
RNAi effects last up to 5 days following introduction of dsRNA. In organisms that have a short life-cycle duration such as Caenorhabditis elegans, the effect can be “inherited” over 1–2 generations. Good results can also be achieved when, instead of a long dsRNA that reflects the length of the original mRNA, straightforwardly short siRNAs are introduced from the outset. Problematic with the method are difficulties to recognize and control non-specific inhibitory effects.
RNAi Technology Enables Thousands of Parallel and Simultaneous Experiments on a Chip and Genome-Wide Screening of Gene Functions. The just outlined method of RNA interference was soon modified yielding several variants, and is still being further developed. For example, thousands of tiny stem cell cultures are placed on a chip and in each single culture one distinct gene is knocked out: Subsequently, all cultures are exposed to a stimulus, for instance to a differentiation factor, that opens the developmental path to, e.g. nerve cells. It is examined whether a given gene is indispensable for the researched process, here for nerve cell development. Automated microscopy facilitates quick evaluation and identification of all those genes needed for the production of a desired cell type. If all gene functions are eliminated, step by step, one speaks of a genome-wide screening, and genome-wide RNAi libraries.
Overexpression or Ectopic Expression After Micro-Injection of Synthetic, Capped mRNA. We ask, whether a gene, the expression of which is subjected to spatial and temporal variation, is of significance for the control of development, or whether its expression is a mere consequence and sign of progressing differentiation. Selected species of cDNAs derived from embryos, or synthetic mRNA, are injected into cells of a host embryo, for example into a cell (blastomere) of the 4- or 8-cell stage, to study its effect. For injection a site is selected where this mRNA normally does not occur (ectopic expression). Or by injection of additional mRNA at the normal site an overexpression is evoked. In embryos of the clawed frog RNA injection has been routinely applied to study gene function.
It even proved successful to begin with mixtures of mRNA that were produced synthetically from pooled cDNA clones. When an interesting effect was observed through repeated subdivision of the mixture (sib selection) eventually the effective mRNA could be isolated. In analogous manner, effects of mixtures of proteins, manufactured in expression systems using mRNA, were checked. Using such strategies the induction factors NOGGIN and DICKKOPF were detected.
Targeted Introduction and Excision of a Gene: The Cre/loxP System
Cre-Lox recombination is a site-specific recombinase technology widely used to carry out deletions, insertions, or translocations of genes. It allows insertion of a (trans)gene to be targeted to a specific organ, tissue or cell type; some modifications of the procedure in addition allow recombination to be triggered by a specific external stimulus. And eventually the tool allows the excision of a previously inserted gene, often the neomycin resistance gene neo, if temporal restriction of its function is intended (in the case of neo to restrict its function to the selection procedure performed to isolate successfully transformed cells or individuals).
A selected foreign gene, for example the gene for the Green-Fluorescing Protein GFP (Fig. 12.7) is in the lab flanked by bacteriophage-derived loxP sequences whereby both lox sequences are oriented in the same direction (only with this orientation the gene can later be excised). Then the construct is introduced into cells, typically into stem cells (the procedure to introduce genes into mice is intricate and will be outlined in Chap. 13). By inserting flanking target sequences corresponding to the gene to be exchanged, the construct is suited to be integrated into the genome by homologous recombination (see Fig. 12.8). However, this happens seldom. To pick out from a cell culture those few cells which have successfully integrated constructs, the whole culture is treated with the antibiotic neomycin. Only cells having taken up and expressed the neo gene survive the treatment. Subsequently a Cre recombinase is introduced into these transformed and selected cells. Introduction is performed either in the form of Cre recombinase protein enclosed in liposomes, or in form of its gene enclosed in a viral vector. Now the loxP site-specific recombinase can remove the neomycin gene out of the genome by a complex recombination event occurring between the loxP sites (Fig. 12.7) (In a first approach one may imagine that the double stranded DNA is cut at both loxP sites by the Cre protein, and the strands are then rejoined with DNA ligase). In Fig. 12.7 a case is shown where, after the excision of the neo selection gene, a reporter gene, be it Lac-Z or a GFP gene, is transcribed. The reporter protein bears witness to the success of the endeavour. One can easily conceive various variants of the procedure to achieve one’s own goals. Placing Lox sequences appropriately allows genes to be activated, repressed, or exchanged for other genes. The activity of the Cre enzyme can be controlled so that it is expressed in a particular cell type or triggered by an external stimulus like a hormone or a heat shock. In elegant variants of the system a cell type-specific promoter is positioned upstream of the recombinase gene. Only in this cell type recombinase will be expressed, and only in those tissues composed of this cell type the neo gene is excised enabling transcription of the downstream reporter gene and any further transgene. In Fig. 13.7 (Chap. 13) a mouse is depicted with a heart-specific promoter placed upstream of a GFP reporter gene, causing the heart of the mouse to fluoresce green. Targeted DNA changes are useful in cell lineage tracing and can provide a first survey of whether a selected gene is required or not for a certain differentiation program or functional task.
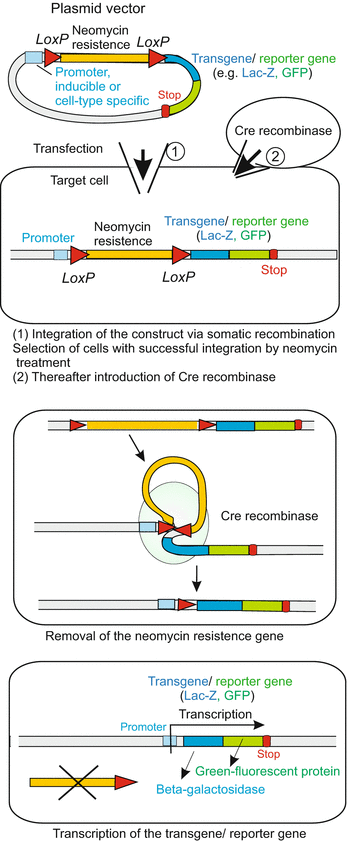
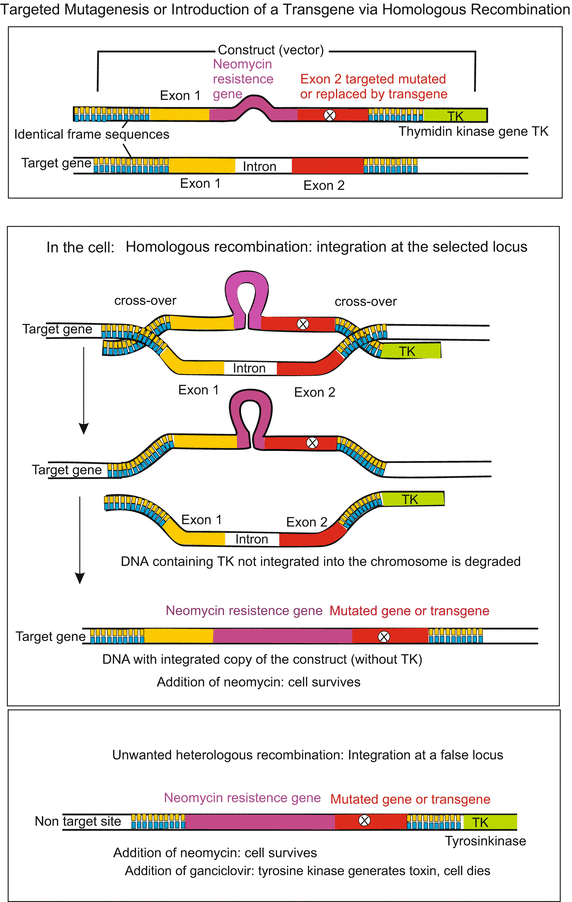
Fig. 12.7
Cre/Lox-System. In the laboratory a construct (vector) is generated which contains the target gene framed by LoxP sequences oriented in the same direction. In addition, the construct can harbour a transgene fused with a GFP reporter gene. Using such vectors transgene LoxP-mice can be prepared (method see Chap. 13). Another group of mice gets a Cre-recombinase gene placed under the control of a tissue-specific promoter. These two groups of transgenic mice are crossed. In the offspring it can happen that the LoxP-framed gene is removed by the activity of the recombinase whereas the transgene-GFP fusion gene remains, is activated through the tissue-specific promoter and causes that tissue to fluoresce green
Fig 12.8
Homologous recombination is used in gene targeting, a technique for introducing genetic changes into target organisms. When an investigator wants to replace one allele with an engineered construct but not affect any other locus in the genome, then the method of choice is homologous recombination. To perform homologous recombination, the DNA sequence of the gene to be replaced must be known. With this information, it is possible to replace any gene with a DNA construct of choice. In the technique frequently used in developmental biology an allele with a targeted mutation or a transgene (foreign gene) is first introduced into stem cells through a plasmid-type construct. This construct may contain, besides the target DNA sequence, additional genes (e.g. for antibiotic resistance, or green fluorescent protein GFP). In any case the construct must include some flanking DNA that is identical in sequence to the targeted locus. The construct is introduced in stem cells through injection or electroporation. By mechanisms that are poorly understood but are similar to what occurs during meiosis and mitosis when homologous chromosomes align along the metaphase plane to repair strand breaks, the engineered construct finds the targeted gene and recombination takes place within the homologous (meaning identical in this case) sequences. The recombination may take place anywhere within the flanking DNA sequences. In the example shown here, the construct contains besides the gene to be exchanged two genes for selection of those few cells in which homologous recombination took place: (a) a neomycin resistance gene enabling survival of only those cells that have integrated the construct and express resistance when the cell culture is treated with neomycin, and (b) a thymidine kinase (TK)-gene, that converts the base-analogous ganciclovir into a toxin. Provided recombination took place at the correct locus, the TK sequence is left over and degraded. If, however, the targeting vector aligns in a non-homologous region of the genome, then recombination is random and the negative selection marker may become incorporated into the genome. Those cells die after administration of ganciclovir. Stem cells that responded according to desire can be introduced into mouse blastocysts where they are expected to give rise to germ cells. These can carry the genetic modification to descendants. Yet, as a rule only one of two alleles will be exchanged by the introduced construct. By repeated inbreeding in the long term also homozygous offspring are to be expected
How Genes Can Be Exchanged for Mutated Variants, and Foreign Genes Introduced
The aim of such experiments is to introduce gene variants, be they natural alleles, foreign genes or artificially designed constructs, permanently into the genome to study their effects in all developmental stages and in following generations (today, preferentially inducible genes or constructs are introduced, see below). How does one proceed?
We already mentioned several technical means to introduce genetic materials into cells, be these materials mutated versions of a gene, foreign genes (transgenes), or artificial constructs, be they naked or enclosed in carriers, the basic techniques are the same: direct injection with micropipettes, electroporation, liposome-mediated uptake, virus-mediated transfer. One common problem remains: Without particular arrangements and designs introduced DNA inserts, if at all, anywhere in the genome and the effects are hard to predict and to control.
Non-Random Integration by Transposons. We used transposons to evoke mutations or as vectors to introduce transgenes into primordial germ cells of Drosophila (Fig. 12.2). The integration of the introduced DNA into the host genome was mediated by transposase introduced by another carrier. But in those days large-scale mutations were carried out for large-scale genetic mapping of developmentally significant genes, it was not possible to determine the locus of integration; the loci were random – almost but not completely. Experience taught that transposons preferentially haunt loci on chromosomes with active genes, perhaps because the chromatin is relaxed there. If by chance a transposon is integrated downstream of the control region of a gene but upstream of the gene proper, an introduced foreign gene can be expressed in a tissue- and stage-specific manner. This applied to the experiment in which supernumerary eyes in flies were successfully induced (see Fig. 12.19).
Targeted Exchange by Homologous Recombination. In general terms, homologous recombination is a type of genetic recombination in which nucleotide sequences are exchanged between two adjacent similar or identical molecules of DNA. It is used by cells during mitosis to accurately repair harmful breaks that frequently occur on both strands of DNA, known as double-strand breaks. We focus on practical aspects and the use of the principle in developmental biology.
In the pioneer model mouse it is possible to render a gene useless at a researcher’s or customer’s option (targeted mutagenesis), or to exchange a gene for another one. The method is based on the rare event of homologous recombination between the introduced and the endogenous gene. The gene vector is linearised, the introduced gene to be exchanged is flanked by sequences which are exactly identical with a large part of the initial and the terminal sequence of the endogenous gene. The procedure for targeted manipulation of mice will be described in Chap. 13 (Fig. 13.6). Here we focus on the procedure in general as it can be carried out in cell cultures (Fig. 12.8). Cells are stimulated to proliferate in the presence of the introduced construct. Occasionally, proliferating cells accomplish exchange following DNA replication but before mitosis using the introduced DNA stand as template for repair of breaks. In order to facilitate selection of those few cells in which recombination took place, a selection marker is included in the construct. When neomycin is present in the culture medium also those cell survive which have integrated the construct and express the resistance gene properly.
In combination with the Cre/loxP system homologous recombination enables excision of a gene only in a particular tissue or organ. After integration of the construct and selection, the neomycin resistance gene (or any other gene) can be removed if this was flanked by loxP sequences. For that a Cre recombinase must be introduced with any vector (see Fig. 13.7).
Methods such as ZFN, TALENs, and CRISPR which are not specifically, or not primarily, developed for analysing developmental processes but to alter the genetic makeup of an organism and to produce transgenic animals are described in Chap. 13.
Permanent Labelling of Living Cells and Their Descendants Using Reporter Genes
There are many reasons to permanently label living cells, for example to track cell genealogies, to follow migrating cells and outgrowing nerve fibres, and identifying them even when they enter and infiltrate a dense network of fibres.
Labelling with dyes is transient even if the dye itself is stable, because with each cell division the concentration of the dye is halved. Not so with reporter genes that are introduced (by injection, electroporation or lipofection). Well-designed constructs are replicated and therefore not diluted out, and are expressed. Skillfully designed constructs have a promoter taken from a cell type-, tissue-, or stage-specific gene and placed upstream of the reporter gene. Often used reporter genes are
β-Galactosidase gene lacZ, of bacterial origin but linked to a eukaryotic promoter (see Fig. 12.11, photo 2), and the
luciferase gene.
After following expression of these genes and addition of appropriate ingredients the presence of these enzymes is detected by the emergence of a blue dye or by emission of light.
Even more impressive and used in innumerable applications is the
GFP (Green-Fluorescent Protein). The gene of this 238 amino acid protein and related genes were isolated from bioluminescent Cnidaria, the prototype from the pacific medusa Aequorea victoria. The protein encoded by this gene spontaneously forms a chromophoric group by rearrangement of three amino acids. The chromophore absorbs excitation light in the blue to ultraviolet range and emits green fluorescent light. The particular advantage is that no separate chromophore or other ingredients must be introduced into the living cells. Once introduced into an organism it is maintained in their genome through breeding.
The particular pleasure is that the glow of the GFP can be seen in the living animal, provided it is transparent (or the light emitting molecules are near the surface, see Fig. 13.5). Under the microscope migrating cells and growing nerve fibres can be observed live and their behaviour documented with a video-recorder.
Research all over the world derives most benefit from the accidental circumstance that the GFP gene, or part of it, can be coupled to a cell’s own gene and expressed with it in the form of a fusion protein, also called a tagged protein (see Fig. 12.11, picture 3, 4, 6; and Fig. 13.5). Astonishingly often, the cell’s own protein retains functionality. Another possibility is to place the GFP gene under the control of a selected cell-type-specific promoter. Expression of the GFP gene is controlled in parallel to the cell’s own (unattached) gene, they are switched on and off simultaneously. In addition, by chemical mutation and modification several GFP variants were made available fluorescing in various nuances and intensities of blue, green, yellow and red, so that under changing excitation waves varying patterns of coloured structures can be observed, each colour representing a distinct gene product.
Inducible Promoters: How Genes Can Be Switched On and Off at the Will of Researchers
Experimental interventions in processes of gene expression are possible when the introduced gene is placed under the control of an inducible promoter. As their name suggests, the activity of these promoters is induced by the presence or absence of an externally added chemical or physical factors. Inducible promoters are very powerful tools in genetic engineering because the expression of genes controlled by them can be turned on or off at certain stages of development of an organism or in a particular tissue.
We refer, for example, to a promoter that can be turned on by the well-known WNT signalling system (Fig. 12.9a). Or to the promoter of the heat shock gene hsp-70, or to the promoter of a steroid hormone receptor gene (Fig. 12.9b). When installed in a cell, the gene controlled via such an inducible promoter can be switched on by a WNT signal, a heat shock, or a steroid hormone.
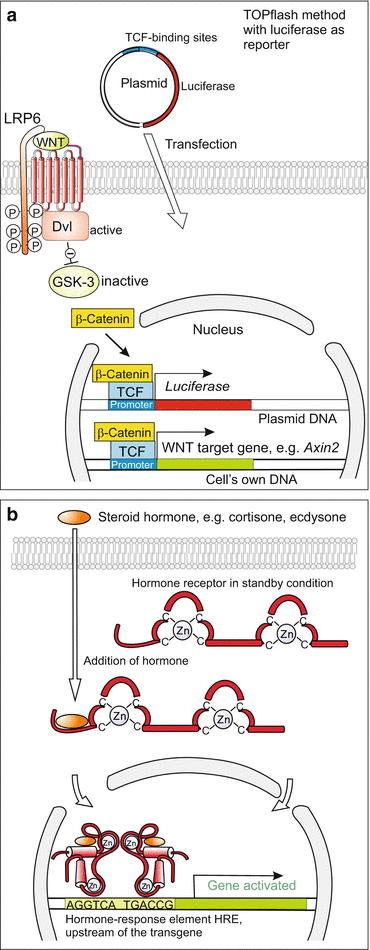
Fig. 12.9
(a) Luciferase-based reporter gene. In parallel to the natural target genes of the WNT signal the introduced luciferase gene is activated as well, since it is under the control of the same promoter. After addition of luciferin substrate the reaction of the substrate with the expressed luciferase produces light signals. (b) Inducible expression systems placed under the control of steroid hormones. A promoter (hormone-responsive element HRE) is set ahead the transgene to be activated. The HRE binds intracellular receptors laden with steroids, and thus the transgene is activated
Since, however, heat shock or a steroid hormone may switch on also other genes of the cell, more specific solutions are required.
The promoter of a mammalian steroid-dependent gene can be substituted by the promoter of the insect-specific steroid hormone ecdysone. Now this gene, and only this gene, can be activated by administering ecdysone).
Many laboratories strive to design more types of artificial promoters that can be handled at the will of the researcher. Example of successful constructs are sophisticated chimeric constructs composed of prokaryotic (bacterial and/or viral) and eukaryotic sequences such as the Tetracycline Response Element (TRE) used in tetracycline-controlled transcriptional activation, also known as tetracycline resistance system, in short Tet system. This is a method of inducible gene expression where transcription is reversibly turned on or off in the presence of the antibiotic tetracycline or one of its more stable derivatives, actually doxycycline Dox. Two variants of the system, named Tet-Off and Tet-On are currently the most commonly used inducible expression systems for research of eukaryote cell biology. Tet-Off activates expression of downstream genes in the absence of Dox, whereas Tet-On and Tet-On Advanced activate expression in the presence of Dox. The benefits of the system are (a) an inducible on or off promoter. The investigators can choose to have the promoter always activated until Tet is added or always inactivated until Tet is added. (b) The second benefit is the ability to regulate the strength of the promoter. The more Tet added, the stronger the effect is. Thus the Tet Technology provides efficient, precise and reversible control over both timing and level of gene expression in eukaryotic cells.
Currently more and more inducible systems are being developed. For instance the following:
Optogenetics: Switching Functions On and Off with Light Impulses in Living Cells and Animals
The term optogenetics refers to newly-introduced technologies to localize and regulate cellular components and to switch functions on and off in milliseconds in living cells or animals that have previously been prepared with genetic technologies. Optogenetic tools allow high-resolution spatial and temporal manipulation of individual cells or subcellular parts such as single synapses. Pioneering experiments were designed to localize synapses equipped with distinct ion channels (Fig. 12.10a,b) and to switch the electric signals emanating at synapses on and off on demand. Through genetic technologies the cells or animals were provided with channel rhodopsins of microbial origin. These are light-gated ion channels that contain retinal and combine sensitivity for light pulses of a certain wavelength with selectivity for either cations or anions (Fig. 12.10b). At present the technology is mainly used in neurosciences to analyse and control neuronal activities in cell cultures, brain slices and in living animals. For controlling the behaviour of freely-moving rodents extremely fine optical fibres are introduced in selected parts of the brain.
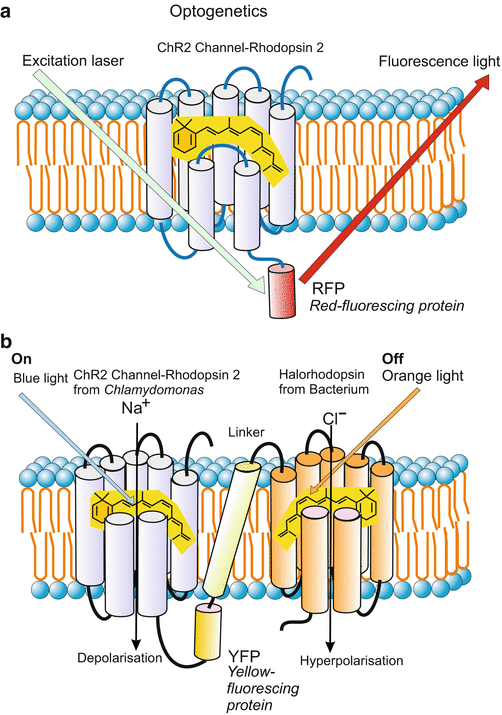

Fig. 12.10
Light-based technologies and optogenetics. (a) Light pulses localize a distinct ion channel to which a derivative of the GFP has been added. (b) On-off switch in the synapse (postsynaptic membrane) of a neuron. The switch consists of a complex of two different photosensitive ion channels of the rhodopsin-channel family coupled with a linker. The ion channel derived from the unicellular alga Chlamydomonas is permeable for cations; blue light induced opening of the channel causes depolarization of the electric membrane potential and thus stimulation of the neuron to fire action potentials. The channel derived from a halobacterium is selective for chloride anions. Its opening through orange light causes hyperpolarization and thus silencing of the neuron. The channels were introduced in human neuronal cell cultures or embryos of rodents by inserting the microbial genes into the genome of the host and placing it under the promoter of specific neurons. After Kleinlogel et al. (2011) as regards content. Own composition by WM
Fig. 12.11




Methods of molecular and cell biology frequently used in developmental biology. (1A) In-situ hybridisation. By means of a labelled antisense probe the presence of an mRNA for a neuropeptide in nerve cells is visualized. All those cells are visible which contain mRNA for neuropeptides of the RFamide (arginine-phenylalanine-amide) class. The object shown is a whole-mount preparation of a polyp of the hydrozoan Hydractinia echinata. The shot shows a detail from the mouth region. Preparation by Prof. Günter Plickert, University of Cologne. (1B) Immunocytochemistry. RFamide nerve cells in the mouth region of a polyp of Hydractinia echinata are visualized using antibodies that recognize RFamide neuropeptides with the C-terminal end arginine-phenylalanine-amide. The antibodies were provided by Dr. C. Grimmelikhuijzen. The antibodies on their part are bound to a secondary antibody coupled with a fluorescing dye. Preparation by the author WM of this book. (2A,B) Reporter gene and targeted mutagenesis for examining how axons of smell cells find the correct glomeruli, i.e. the first stations of data processing in the bulbus olfactorius (olfactory brain). These axons contain in their cell membrane smell receptors identical to those present in the cilia of differentiated smell cells and used there for odour detection. In this experiment the hypothesis was examined that the molecular smell receptors in the cell membrane along the axons contribute to target finding. In a sophisticated multi-step procedure first in mice the gene for such a receptor was exchanged by a mutated gene using the method of homologous recombination. The mutated gene was linked with a neomycin resistance gene framed by LoxP sequences. Subsequently, the neomycin cassette was removed using Cre-mediated recombination (method similar to that shown in Fig. 12.7) and exchanged for a LacZ construct. When the introduced LacZ gene is expressed the presence of the LacZ protein (=bacterial β-galactosidase) can be visualized by means of a dye reaction. 2A Shows growing axons, 2B shows how these axons form synapses in the target area of the brain. Result of the experiment: A single exchange of one amino acid can suffice to guide the bundled axons to another target area (after Feinstein and Mombaerts 2004, Feinstein et al. 2004). The photos were kindly provided by Prof. Peter Mombaerts, Rockefeller University New York. (3–4) Fluorescing tags. (3) Brain of a mouse with blue and green fluorescing neurons. The mouse had inherited a gene for GFP (Green Fluorescent Protein) from its father and a gene for CFP (Cyan Fluorescent Protein) from its mother. Image copied with kind permission of Dr. Jeff W Lichtman, Washington University, School of Medicine (rights reserved). (4) Purkinje-neurons in the cerebellum of the mouse, arisen from the spontaneous fusion of a GFP stem cell with a nerve cell. Image kindly provided by Dr. Clas B. Johansson, Karolinska University Hospital Stockholm, with permission of Prof. Dr. Helen Blau, Stanford. For method see Johansson CB et al. (2008) Nature Cell Biol 10(5): 578–83. (5) Embryo of Drosophila at the blastoderm stage. Threefold immuno-fluorescence with antobodies to HAIRY protein in red, to KRÜPPEL in green, and to GIANT in blue, Laser scanning-confocal microscopy. Image kindly provided by Dr. Stephen Paddock, Lab. Cell & Mol. Biol., Wisconsin University. (6) Bright heart of Drosophila with luminescent GFP. The cells of the heart of living embryos of Drosophila were made fluorescent by placing the reporter GFP gene under the control of the hand promoter. The hand gene codes for a transcription factor of the bHLH protein family highly conserved in the animal kingdom. The promoter of the hand gene is switched on in the cardiomyoblasts, in the heart-associated pericardial cells, and in the cells of lymph glands. When GFP is matured the heartbeats can be observed live as well as the further development of the heart. The image was kindly provided by Prof. Dr. Achim Paululat and Julia Sellin, University of Osnabrück
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
