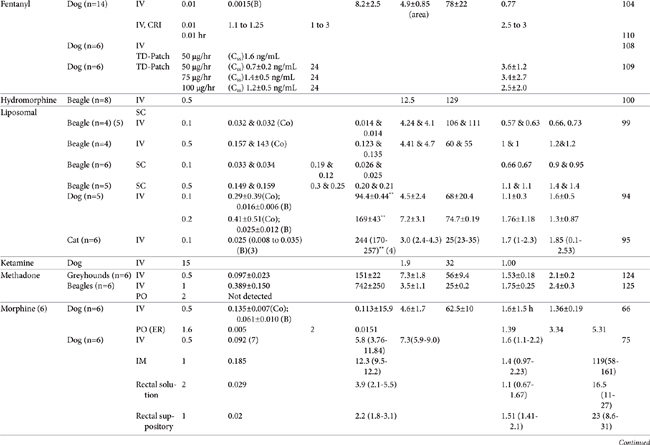
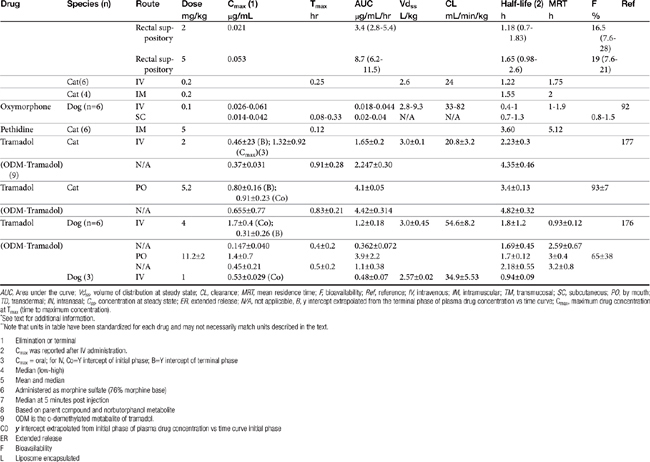
AUC, Area under the curve; Vdss, volume of distribution at steady state; CL, clearance; MRT, mean residence time; F, bioavailability; Ref, reference; IV, intravenous; IM, intramuscular; TM, transmucosal; SC, subcutaneous; PO, by mouth; TD, transdermal; IN, intranasal; Css, concentration at steady state; ER, extended release; N/A, not applicable, B, y intercept extrapolated from the terminal phase of plasma drug concentration vs time curve; Cmax, maximum drug concentration at Tmax (time to maximum concentration).
∗ See text for additional information.
∗∗ Note that units in table have been standardized for each drug and may not necessarily match units described in the text.
| 1 | Elimination or terminal |
| 2 | Cmax was reported after IV administration. |
| 3 | Cmax = oral; for IV, Co=Y intercept of initial phase; B=Y intercept of terminal phase |
| 4 | Median (low-high) |
| 5 | Mean and median |
| 6 | Administered as morphine sulfate (76% morphine base) |
| 7 | Median at 5 minutes post injection |
| 8 | Based on parent compound and norbutorphanol metabolite |
| 9 | ODM is the o-demethylated metabalite of tramadol. |
| C0 | y intercept extrapolated from initial phase of plasma drug concentration vs time curve initial phase |
| ER | Extended release |
| F | Bioavailability |
| L | Liposome encapsulated |
Chapter 28 Control of Pain in Small Animals: Opioid Agonists and Antagonists and Other Locally and Centrally Acting Analgesics
Definition of Pain and its Recognition
Classification of Pain
The International Association for the Study of Pain (IASP) defines pain as an unpleasant sensory and emotional experience associated with actual or potential tissue damage.1 During the past several decades, the importance of pain has evolved from being underestimated, to being recognized for its contributions to morbidity and mortality. In 2000 the Joint Commission on Accreditation of Healthcare Organizations2 mandated that hospitals for humans consider pain as the fifth vital sign, thus ensuring its inclusion in the routine evaluation of all patients.
Pain has been classified in several ways. Severity (mild, moderate, and severe) is among the most common, but this categorization also includes an emotional component of pain. As such, in veterinary medicine this classification requires interpretation by either the clinician or owner. Pain also is classified according to its source. Physiologic pain is a protective mechanism, leading the animal to withdraw from a potentially damaging stimulus. In contrast, pathologic pain reflects tissue damage. Unresolved physiologic pain eventually can lead to pathologic pain by causing neurologic damage (also classified as neuropathic pain). Neuropathic pain is also classified according to the location at which the damage occurs (e.g., neurologic damage), with nociceptive pain reflecting tissue damage. Pain might also be localized to the body site: visceral pain is associated with abdominal or thoracic pain, whereas somatic pain originates from musculoskeletal damage. Either of these locations can be further divided as superficial or deep. Pain also is described by duration. Acute pain is abrupt in onset and resolves in 24 to 72 hours, whereas chronic pain is slow in onset and generally persists for several weeks to months. Acute pain is more protective in nature (compared with chronic pain) and indicates that something is going wrong, whereas chronic pain reflects disturbed homeostasis. Acute pain results from traumatic, surgical, or infectious events and generally is conducive to resolution with analgesics. In contrast, chronic pain is a long-standing physical disorder or emotional distress and includes pain associated with degenerative joint disease and some types of cancer. Chronic pain may not respond to traditional analgesic therapy, with tranquilizers or behavior-modifying drugs combined with environmental manipulation and behavioral conditioning commonly implemented. The transmission of chronic pain increasingly is being understood, with knowledge regarding neural pathways elucidating possible targets of therapy. Accordingly, newer drugs are increasingly being identified for their potential efficacy.3
Transmission of Pain
Nociceptive Pain
Nociception refers to a neural response of nociceptors to a noxious stimulus.1 Nociception is a normal nervous system function that serves as a warning of danger or credible threat. The nociceptive response comprises a nociceptor and three neuron chains, originating in the peripheral tissues and ending in the cerebral cortex. Nociception is unique among sensory nerves because nocicpetors must detect a wide range of stimuli, including physical and chemical signals, heat and cold, and acid and mechanical pressure. Nociceptors are located in every tissue of the body, originating from a group of neuronal bodies in the dorsal root ganglia (Figure 28-1). Nociception varies with location (peripheral versus central) and organ. Nociception in the skin occurs at the surface (i.e., is somatic), resulting in sharp, defined, localized, and limited pain. In contrast, internal, or visceral, nociception is generally diffuse, dull, and often referred.4 The action potential stimulated by a nociceptor crosses the synapse in the dorsal horn and stimulates the second order neurons in the gray matter of the spinal cord. There the signal is transmitted to the brain, where it is processed and interpreted. The transmission of pain from nociceptors is carried by either small, myelinated A delta fibers, smaller unmylelinated C fibers, or A a fibers. A delta fibers are fast, being responsible for sharp and acute pain, and transmit somatic and parietal pain. Because these receptors are discrete, animals can localize this pain. In contrast, C fibers are slow, transmitting dull, aching, burning, or throbbing pain that is difficult to localize. A fibers also are slow, transmitting stimuli associated with vibration, stinging, or tickling. However, because they are not able to discriminate between painful versus nonpainful stimuli, they are not pure nociceptor neurons.
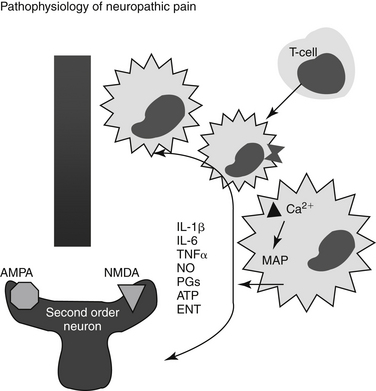
Chemical mediators are important components of the nociceptor reflex and offer a target of pharmacologic modulation. These include but are not limited to adrenocorticotropic hormone, glucocorticoids, vasopressin, oxytocin, brain opioids, catecholamines, angiotensin II, endorphins/enkephalins, vasoactive intestinal peptides, substance P (centrally), eicosanoids (prostaglandins, leukotrienes), tissue kininogens (bradykinin), histamine, serotonin, potassium, and proteolytic enzymes.6 Several of these mediators are also associated with stress. Several neurotransmitters associated with nociception and transmission of peripheral pain also function in the dorsal horn, the first site of signal processing. Spinal neurotransmitters include but are not limited to peptides (e.g., substance P, calcitonin gene-related peptide), excitatory and inhibitory amino acids (aspartate, glutamate, gamma-aminobutyric acid [GABA]), nitric oxide, prostaglandins, adenosine-5’-triphosphate (ATP), endogenous opioids, monoamines (serotonin, norepinephrine), protons (acids), and neurotrophins. Opioid peptides are synthesized by interneurons in the superficial dorsal horn; they regulate further neurotransmitter release, probably through decreased calcium conduction. Glutamate, the primary excitatory nociceptor neurotransmitter, is released in response to calcium, following depolarization. Subsequent postsynaptic binding to N-methyl-d-aspartate (NMDA) receptors influences pain transmission, hyperalgesia, allodynia, and neurotoxicity, thus providing the rationale for combination analgesic therapy that includes NMDA-receptor antagonists.
The IASP definition of pain includes both a sensory and emotional component.2 Indeed, of all the sensory systems, the nociceptive system is most able to elicit an arousal response in the brain. The emotional portion of pain occurs at the level of the limbic system (emotional), cortex (cognitive appraisal), and frontal lobes. Stimulation in the reticular formation results in emotional reaction to pain (anxiety, depression, suffering), whereas stimulation in the cerebral cortex leads to conscious perception and interpretation of pain. Transmission of central pain reflects a “gate control” phenomenon. The dorsal horn cells modulate the patterns of incoming information transmitted to the brain, which ultimately produces response and perception. The brain, in turn, can enhance or counter nociception. For example, when pain is sustained, released opioid peptides bind mu receptors in the brain and spinal cord, decreasing pain. Modulation regulates pain response, thus maintaining homeostasis.7 In addition to central transmission of pain, the first-order neuron can also synapse with neurons that cause a local reflex. The reflex can be myoneural or sympathetic in action (e.g., release of norepinephrine, smooth muscle spasm, vasoconstriction). Voluntary reflexes require conscious pain perception, whereas nociceptive reflexes do not.
Not surprisingly, a variety of mechanisms transduce nociceptive signals.8 At least two classes of channels are involved: One class detects noxious stimuli or products of tissue damage, and another sets the threshold necessary for a nociceptor action potential. Channels that detect noxious stimuli include transient receptor potential (TRP) channels, acid-sensing ion channels (ASICs), and the ATP-gated P2X receptor family. Of these, the TRP channels are emerging as the dominant channels transmitting nociceptor signals. The TRP channels are general receptors, able to sense multiple types of noxious stimuli. Their actions are complex, with channels having different roles in different tissues. Further, each channel is able to respond to more than one stimulus and with overlapping sensitivities. Four of the nine currently described nociceptive TRP channels respond to heat. The TRP receptors are regulated through a number of kinases (e.g., kinase C, kinase A, calcium/calmodulin–dependent kinase). In contrast to the general TRP channels, ASIC channels are specialists, able to detect only acidity, as might occur with bone cancer (high levels of acid leak into surrounding tissues), inflammation (local pH can drop to 5.5), and ischemia (resulting from metabolites such as lactic acid). Because extracellular ATP is a major product of inflammation, P2X channels are also drawing interest for their potential role in inflammatory pain, particularly visceral pain.7
Neuropathic Pain
Pathophysiology
Nociception is not limited simply to transmission of acute (nociceptive) pain; it also contributes to neuropathic pain. Both acute and unrelieved chronic pain can shift from nociceptive to neuropathic pain. Neuropathic pain includes hyperalgesia (overreaction or increased sensitivity to painful stimuli), allodynia (reaction to an innocuous stimulation), or reflex sympathetic dystrophy. The latter is a complex disorder of pain, sensory abnormalities, abnormal blood flow, sweating, and trophic changes in superficial and deep tissues.7 Failure to control development of acute pain and hyperalgesia can lead to chronic pain; progressive and prolonged stimulation can lead to a “wind-up” phenomenon that reflects increased excitation of neurons in the dorsal horn. The wind-up phenomenon is manifested as a pain response outside the site of injury and can persist beyond resolution of the inciting cause (pathologic pain). Dysesthesia is another example of neuropathic pain characterized by an unpleasant spontaneous or provoked sensation. Dysesthesia has been described in humans as a burning or shooting sensation reflecting damage to peripheral nerves. Excess stimulation of pain fibers, or reduced activity of non-nociceptive sensory pathways contribute to an imbalance between painful and nonpainful inputs to the central nervous system (CNS).9 Examples include phantom pain, a centrally mediated pain manifested as an intense burning sensation at the nerve ending of a damaged, paralyzed, or missing extremity. Another example is “stump” pain, a peripheral neuropathic pain secondary to neuroma formation at the site of amputation. These are examples of pain without any obvious noxious input. Further, for some chronic or persistent pain disorders, pain does not necessarily originate at the periphery.4
Heightened pain sensitivity is probably a protective response, reducing tissue exposure to further risk of tissue damage, and normally resolves when the tissue is healed. Both local and spinal changes contribute to neuropathic pain. Tissue damage intensifies the sensation of pain by recruitment of otherwise silent receptors. Chemical mediators are produced and released by both neuronal and non-neuronal cells (e.g., fibroblasts, mast cells, neutrophils), increasing nociceptor sensitivity. Local injury causes release of inflammatory mediators (cyclooxygenase-2 [COX-2], interleukins [ILs], leukotrienes [LTs], H+, K+, histamine, bradykinin), generating a slightly acidic inflammatory milieu that lowers threshold sensitivity (allodynia) by facilitating chemical binding to nociceptors. Activation of the nociceptors initiates neurogenic inflammation, leading to the release of substance P and peptides, which in turn perpetuate inflammation.5 Dysesthesia may reflect spontaneous activity originating in the regenerating, primary, nociceptive neurons (myelinated, small afferent fibers). Changes in dorsal root Na+ channels also have been reported. Dysesthesia may thus reflect a focal inflammatory process as opposed to axonal damage. Further, potential involvement of (fast) Na+ channels that generate ectopic discharges supports the use of local anesthetics (Na+ channel blockers) as part of a combined analgesic approach.9
Heightened sensitivity also involves retrograde signals from the site of injury to the neuronal cell body; signals alter neurotransmitter release and receptor transcription and expression. Among the mediators of hypersensitivity are mitogen-activated protein kinases (MAP: p38, ERK and JNK), which may initiate changes in the microglia of the dorsal horn.6 These changes may persist for weeks. Other mediators released and associated with hypersensitivity in the spinal cord include but are not limited to the proinflammatory cytokines tumor necrosis factor (TNF), IL-1 and IL-6, prostaglandins, and reactive oxygen species. Microglial cell bodies also hypertrophy and multiply, a reaction that, along with release of proinflammatory cytokines, may be inhibited by minocycline. Interestingly, chronic morphine administration may activate microglial cells and thus exacerbate hypersensitivity, an effect that might be reversed by minocycline or pentoxifylline, exemplifying the complexities of hyperalgesia.10
A role for NMDA receptors has not yet been identified in normal spinal cord transmission. However, a role might be suggested in transmission of pain. Both glutamate and aspartate are major excitatory neurotransmitters in the CNS that bind to NMDA receptors. After tissue injury, impulses from sensitized nociceptors of C fibers stimulate glutamate and other chemical release (e.g., neurokinins) in the primary afferent in the spinal cord. Calcium influx and activation of early genes leads to the development and maintenance of hypersensitivity or the wind-up phenomenon and hyperalgesia. Hyperalgesia associated with opioid use may involve NMDA receptors (see later discussion). Another mediator group that may be involved with changes in response to pain is prostaglandins (PGs). Induced COX-2 prostaglandin E (PGE) has been associated with hyperalgesia in either the spinal cord (primary hyperalgesia) or peripherally at nociceptors (secondary hyperalgesia).11 Induction of spinal COX-2 in the dorsal horn has been associated with central sensitization.11–13 Finally, PGs may potentiate the effects of other chemical mediators involved in pain or inflammation (e.g., histamine, bradykinin, substance P, nitric oxide), neurotransmitters (e.g., inhibition of glycine or potentiation of glutamate), or other receptors (e.g., NMDA), although this role in central sensitization and hyperalgesia has yet to be determined.
Nociception may awaken long dormant processes. For example, GABA is normally inhibitory, but it becomes excitatory in the spinal cord after peripheral nerve injury.7 Further, if the inciting cause is sufficient, quiescent nociceptors will be recruited; output of non-nociceptive neurons (e.g., touch receptors) may be interpreted as nociception by second-order neurons in the spinal cord.5
Preemptive control
Meta-analyses of human studies that focused on timing of analgesia in surgical patients warrant review for their implications with regard not only to timing but also to the importance of effective pain control.14,15 One meta-analysis examined randomized, double-blinded, controlled trials that addressed either preemptive or postoperative analgesia for acute or chronic postoperative pain relief. Analgesic protocols that were reviewed studied the following: Twenty of the clinical trials reported the analgesic efficacy of NSAIDs (n=20; diclofenac, ketorolac, ketoprofen, ibuprofen, flurbiprofen, and naproxen [endodontal]), opioids (n=8; morphine, fentanyl, alfentanil, sufentanil, meperidine), and NMDA antagonists (n=8; ketamine, dextromethorphan). Further, epidural, caudal, or intrathecal (n=20) or local anesthetics (n=20) were considered. Outcome measures included comparison of pain scores before and 24 hours after surgery, time to the need for analgesics, and the need for supplemental analgesic therapy. In general, meta-analysis concluded that pre-emptive timing did not alter the magnitude of postoperative pain, although the consumption of analgesia and the time to request additional analgesics were reduced. For NMDA antagonism, preemptive use of ketamine (as part of combination analgesic therapy, generally with opioids) uniformly did not cause a statistical difference in postoperative pain (a finding substantiated by both meta-analyses), whereas dextromethorphan did, although only two trials were reviewed. For epidural analgesia 7 of 11 trials using opioids indicated potential benefits, although the authors concluded that clinical relevance would be improved if epidural analgesia continued for a longer postoperative period. In general, preemptive nonsteroidal antiinflammatory drug (NSAID) therapy provided no difference in pain relief, leading the authors to conclude that the risks associated with preemptive NSAID therapy (e.g., bleeding, renal compromise) may not be justified; the power of the study to detect a significant difference is not known. Only one of the trials addressed the advent of chronic (6 months) pain after a surgical procedure. This trial found chronic pain to be significantly reduced in patients receiving preemptive analgesia. The overall conclusions by the authors was that preemptive analgesia may not be an effective method for reducing postoperative pain. However, the authors also suggested that future studies should not focus on the timing so much as on the approach to prevention of postoperative pain. Specifically, studies might address the importance of aggressive control, including the use of combinations designed to prevent neuropathic pain. Indeed, the lack of evidence of effective preemptive analgesia for control of postoperative pain reported in this study may reflect failed actions that occur in clinical patients. Because even abbreviated sensory events may initiate events leading to central sensitization, effective prevention may be dependent on continuous sensory blockade—that is, throughout the presence of the stimulating nociceptive event. Further, the success of afferent blockade may depend on blockade of input from small nonmyelinated C fibers or spinal, rather than peripheral, analgesia. Indeed, the meta-analysis indicated that neither systemic morphine nor small doses of intrathecal opioids had an effective preemptive impact. However, larger intrathecal doses of morphine were effective. Thorough regional blockade may also be paramount to the success of preemptive operative analgesia.
Pharmacologic Control of Pain
Analgesia reflects the selective interruption of the transmission of injury signals (real or potential) between primary sensory neurons, the spinal cord dorsal horn, the rostroventral medulla, and the cortex opioids.5 The paradigm for the approach of pain control in both human and veterinary medicine has been shifting with the increasing realization that uncontrolled pain is not only bad but also avoidable.16,17 Appropriate pain management is part of the practice of good medicine: For human patients, analgesic control is associated with more rapid clinical recovery, shorter hospital stays, fewer readmits, and improved quality of life.1
Control of Pain
Assessing Response to Pain and Stress
One of the more difficult aspects of acceptable control of pain for the clinician is detection or recognition of pain.19–21 Scientists agree that all animals feel pain, although the level of nociception may vary between vertebrates and invertebrates and among classes of animals.22 Animals may feel pain as easily as human patients; any stimulus that is likely to cause pain in a person will cause pain in animals. Animals differ from people, however, in their response to pain. Indeed, the laws of behavior in wild animals require that abnormal behavior associated with pain be avoided. Avoidance, escape, or control of pain and distress are responses to pain that are important for (wild) animal survival and allow animals to adapt to a new or changed environment. Animals showing weakness, pain, and distress become targets for predators. Ill or injured animals tend to be abandoned by others so that an entire stock is not jeopardized. This evolutionary process makes clinical recognition of pain in animal patients difficult. For example, a critical patient is often unable to manifest pain and unwilling to care for itself; the clinician must be diligent to recognize the likelihood of an underlying disorder in causing pain. Increase in heart rate usually is caused by the underlying pathology, not in response to pain. The more severe the illness, the more important the need for analgesics.
Response to acute and chronic pain varies and includes both physiologic and behavioral changes. If pain is too severe for the animal to accommodate, a state of distress can develop.23 Beyond protection, pain rarely has any useful function and is associated with dramatic and potentially life-threatening physiologic changes.24–26 Physiologic responses to distress include gastrointestinal lesions, immunosuppression, and hypertension. In human patients failure of response to treatment, hospitalization duration, and hospitalization costs can be positively correlated with failure of effective pain control. The sensation of pain can be associated with a marked adrenergic (catecholamine) release, which may cause life-threatening hypertension or cardiac arrhythmias. Response to acute pain includes physiologic changes such as tachycardia, tachypnea, mydriasis, and salivation and behavioral responses such as guarding, protection, vocalization (especially with movement or palpation of painful area), licking, biting or scratching, shaking, restlessness or insomnia, and recumbency.
Failure to control acute pain can stimulate changes at the level of the nociceptor and CNS; nociceptive pain may progress to neuropathic pain. Neuroplastic changes such as hypersensitization or allodynia worsen the stress response that often follows surgery. The response is adaptive and is mediated by behavioral, neural, endocrine, immune, hematologic, and metabolic changes that attempt to restore homeostasis. However, the combined effects can prove detrimental to the patient. Potential sequelae include cardiovascular instability, hypercoagulability, insulin resistance, increased metabolic rate and protein catabolism, and immunosuppression. As such, effective control of pain is paramount in the postoperative or otherwise stressed patient.
Variability among animals in response to stress also makes diagnosis of pain difficult (Box 28-1).
Box 28-1 Types of Biologic Response to Pain
Types of biologic response to pain vary with genetic, age, or physiologic state or makeup:
The American Veterinary Medical Association reviewed the major consensus concepts generated by the Cross-species Approach to Pain and Analgesia workshop that took place in 2002.22 In addition to recommendations regarding the use of animals in research involving pain, the review provides guidelines for development of a pain assessment tool. The guidelines stipulate that a numeric (1-10) scale, based on observable behaviors and quantifiable biological markers, be used. Factors that are likely to alter assessment include animal factors such as species (and strain), stage of development (including prepartum and postpartum development), gender, previous experience to pain; environmental factors; and the type of pain. Other factors, such as nutrition, drugs, concurrent disease, and owner social status, should also be considered. The American Society of Anesthesiologists developed a pain-scoring system (for humans) based on several categories, each scored from 1 to 15. Included are behavior (depressed = 1, normal = 3, apprehensive = 5, and excited or aggressive = 10), preexisting pain (none = 1, minimal = 5, moderate = 10, and severe = 15), surgically induced trauma or pain (minimal = 5, moderate = 10, and severe = 15), duration of surgery (less than 1 hour = 5, 1 to 2 hours =10, and >3 hours = 15), and patient health (normal = 1, mild disease = 2, severe disease = 3, moribund = 4, and life-threatening = 5).
Endogenous and Exogenous Pain Control
Endogenous opiates (opiopeptins) provide analgesia when released in high concentrations in selected regions of the brain. These include enkephalins, dynorphins, and endorphins (Figure 28-2). Each opiopeptin is derived from a larger precursor molecule. Each of the precursor molecules has a characteristic anatomic distribution that is not limited to the CNS. The precursor for endorphin (β–endorphin) is pro-opiomelanocortin, which is also the precursor of melanocyte-stimulating hormone as well as adrenocorticotropic hormone and beta-lipotropin, suggesting a strong link between the opioid system and stress hormones.27 Endogenous recognition sites for these chemicals are also the targets of the exogenous drugs. A variety of other neuropeptides also have been implicated in endogenous analgesia (e.g., vasopressin, neurotensin, cholecystokinin, substance P). Some of these act in concert with other chemicals to stimulate nociceptors. Most notable are the eicosanoids (PGs, LTs), substance P, and bradykinin. The inflammatory process involves the release of a number of these chemical mediators either from the tissues at the site of infection or from the inflammatory cells themselves. Control of pain caused by these mediators is often largely dependent on controlling the inflammatory process. Nociceptin (NC, orphanin FQ, or OFQ) is a novel endogenous opioid peptide named for its ability to lower some pain thresholds. The NC/OFQ system is non-opioid, characterized by behavioral and pain modulator actions that differ from the classical opioids.27 Nocistatin is a related protein of the NC/OFQ system.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


