CHAPTER 46 Commercial Pet Food–Related Nephrotoxicity
ETIOLOGY
Nephrotoxicants typically lead to acute kidney injury (AKI), although affected cats may develop chronic kidney disease (CKD) if they survive the initial episode. Acute injury is defined as a rapid decrease in the glomerular filtration rate (GFR). Acute injury can be classified into prerenal (functional response of structurally normal kidneys to hypoperfusion); intrinsic renal (involving structural damage to the renal parenchyma); or postrenal (urinary tract obstruction) causes. Glomerular, vascular, tubular, or interstitial injury may predominate in intrinsic injury. The kidneys are particularly susceptible to the effects of toxicants (Box 46-1); the organs receive 20 per cent of the cardiac output, and the renal cortex receives 90 per cent of total renal blood flow. Furthermore, their role in excretion means that various substances are concentrated in the kidneys as a result of tubular secretion or reabsorption.1
The most common mechanism of nephrotoxic damage is intrinsic renal injury caused by acute tubular necrosis (ATN), which leads to reabsorption of the glomerular filtrate. Other potential mechanisms include tubular epithelial damage and formation of casts with subsequent tubular blockage; renal vasoconstriction and ischemic necrosis; and changes in the glomerular ultrafiltration barrier.2 In the context of pet food–related nephrotoxicity, recent episodes have included crystal nephropathy and hypercalcemic nephropathy.
PATHOGENESIS
The role of the proximal convoluted tubule (PCT) in concentrating and reabsorbing the glomerular filtrate renders it vulnerable to toxic injury. Cats may be susceptible to ATN because their small size predisposes them to this dose-dependent phenomenon. Toxic ATN may be caused by a wide variety of substances that potentially could contaminate pet foods, including heavy metals, organic solvents, mycotoxins,3 and multiple classes of drugs.4 The PCT contains enzymes that bioactivate agents (e.g., cytochrome P450); an increased transport of organic anions, cations, and heavy metals leading to their accumulation; and an increased susceptibility to ischemic injury. Toxins may damage the tubular epithelium by several subcellular mechanisms. The parent compound, or metabolites, may impair the normal function of mitochondria, disrupt lysosomal and cell membranes, shift ion gradients (i.e., intracellular calcium), and lead to free radical formation. Epithelial cells are sloughed into the tubular lumen and contribute to cast formation. The casts then increase intratubular pressure and decrease the GFR. Loss of the epithelial barrier and the tight junctions between cells can result in back-leakage of the glomerular filtrate and further reduction in the effective GFR.5
Crystal nephropathy occurs when compounds precipitate in the intratubular lumen. The concentration of toxic crystals may be exacerbated further in cats with preexisting renal disease, in which remaining functional nephrons undergo compensatory solute diuresis. Crystal formation of specific compounds also is favored in the acidic urine typically produced by cats fed proprietary diets. A recent episode of pet food–related nephrotoxicity was attributed to contamination of wheat gluten, rice protein, and corn gluten with melamine and cyanuric acid.6,7 It was presumed that melamine was added intentionally to increase the apparent protein content of the food and monetary value of these ingredients; cyanuric acid may have been a co-contaminant because it was used in melamine synthesis. Individually, melamine and cyanuric acid were relatively nontoxic; however, in combination they led to macroscopic and microscopic crystal deposition within the lumen of distal tubules and collecting ducts, severe interstitial edema, and hemorrhage at the corticomedullary junction (Figure 46-1).8 Exposure to a single oral dose of melamine and cyanuric acid at dietary concentrations of 2000 mg/kg (0.2 per cent) was sufficient to cause acute renal failure. Concentrations of melamine in contaminated pet food samples ranged from 10 to 3200 mg/kg (0.001 to 0.32 per cent).8 A survey performed by the American Association of Veterinary Laboratory Diagnosticians found that 347 animals were affected over a 2-month period; sixty-one per cent of the 235 cats died.
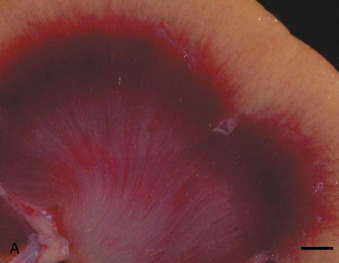
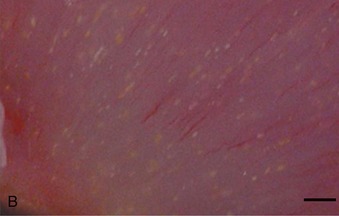
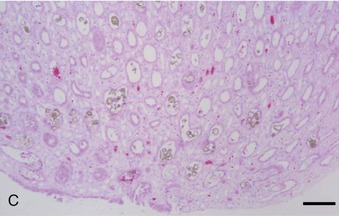
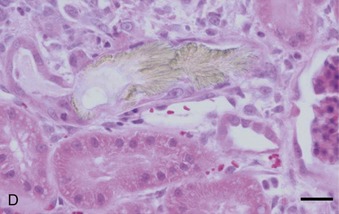
A third example of pet food–related nephrotoxicity is hypervitaminosis D and hypercalcemic nephropathy. Hypervitaminosis D may be caused by excess dietary cholecalciferol (vitamin D3) or ergocalciferol (vitamin D2). Hypercalcemia results from increases in intestinal absorption, osteoclastic bone resorption, and distal renal tubular reabsorption of calcium. It causes decreased urinary concentrating ability and polyuria by decreasing tubular reabsorption of sodium and by impairing the action of antidiuretic hormone on the tubular cells of the collecting duct. Hypercalcemia may cause azotemia by the following multiple mechanisms: prerenal reduction in extracellular fluid volume (anorexia, hypodipsia, vomiting, and polyuria); renal vasoconstriction caused by ionized hypercalcemia; decreased permeability of the glomerulus; and ATN from the ischemic and toxic effects of hypercalcemia (altered cell permeability and calcium pump activity, decreased cellular membrane permeability production and energy production, and cellular necrosis).9 CKD may result from the ensuing nephron loss, nephrocalcinosis, tubulointerstitial inflammation, and interstitial fibrosis.10
All commercial cat foods provide vitamin D in excess of minimal requirements, and there is no regulated upper limit on the amount of vitamin D that may be included. Cats seem to be relatively resistant to cholecalciferol toxicity when the diet is otherwise complete and balanced; the effects of hypervitaminosis D may be modulated by increased dietary calcium and phosphorus and decreased dietary magnesium.11 However, hypervitaminosis D in commercial pet foods has resulted in azotemia, nephrocalcinosis, and progressive renal disease and failure in cats.12–15 Analysis of the diets showed the cholecalciferol concentrations exceeded dietary requirements by 30 to 100 times; renal failure occurred 4 to 14 months later.13 In 2006, an episode of hypervitaminosis D and hypercalcemic nephropathy occurred after incorrect formulation of a mineral mix; the number of animals affected was not disclosed.16
CKD may be associated with long-term exposure to toxins and is mostly related to secondary pathological changes triggered by the initial injury. Different mechanisms are thought to cause the progressive nature of CKD: increases in single-nephron GFR and renal blood flow may promote intraglomerular hyperperfusion and hypertension, and/or systemic hypertension leading to glomerulosclerosis and proteinuria17; hypokalemia may cause tubulointerstitial inflammation and interstitial fibrosis nephropathy; and secondary renal hyperparathyroidism may cause nephrocalcinosis. Ultimately, these chronic changes may progress to an end-stage kidney. The initial insult usually is undetermined.
CLINICAL SIGNS
Clinical signs associated with pet food–related nephrotoxicity depend on the dose of the toxic agent, the presence of associated diseases predisposing to renal injury (Box 46-2), and the severity of AKI. Acute renal dysfunction is characterized by rapid development of depression, anorexia, vomiting, and diarrhea. Nephrotoxicants may cause polyuria (>2 mL/kg/hr), oliguria (<0.27 mL/kg/hr), or anuria (<0.08 mL/kg/hr). Physical examination may demonstrate a good body condition score, dehydration, hypothermia, oral ulceration, uremic breath, tachycardia or bradycardia, and painful renomegaly. Important differential diagnoses for the latter physical findings include ureterolithiasis, renal lymphoma, and other toxicities (e.g., lily intoxication) (Box 46-3).



