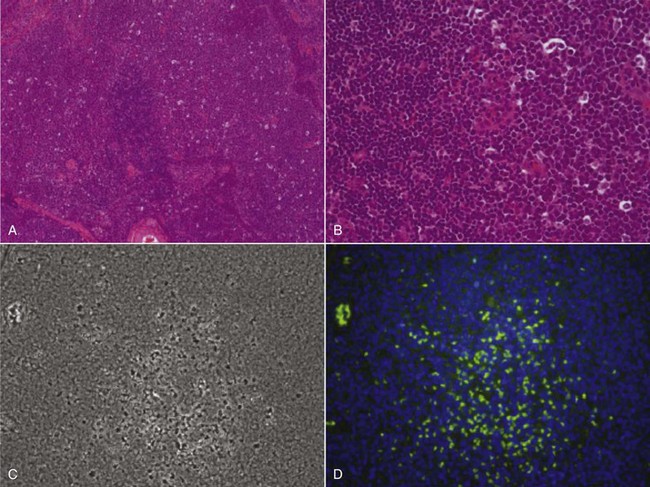
17 Oncology clinical trials attempt to answer questions and find better ways to prevent, diagnose, or treat cancer. Their model is different from trials involving infectious or even chronic diseases because the risks involved have greater morbidity and mortality. Traditional drug development follows a strict, step-wise paradigm that begins with a phase I dose-finding trial, followed by a phase II efficacy/activity trial, and concludes with a phase III “pivotal” trial that pits a novel agent against or with the current standard of care (Table 17-1).1,2 Although veterinary oncology trials sometimes combine these concepts, their individual descriptions serve as the framework for new drug development. Clinical designs, pertinent endpoints and analyses, the process for drug approval, and clinical trial ethics will be explored in the following sections. Table 17-1 Goals of Phase I–III Clinical Trials MTD, Maximum tolerated dose; DLT, dose-limiting toxicity; PK/PD, pharmacokinetic/pharmacodynamic. Phase I trials are the first step in the evaluation of a new agent or biologic. The primary goal is to determine a tolerable dose to be used in future studies by evaluating safety, tolerance, and dose-limiting toxicity (DLT).2–4 Typically, safety is determined in increasing dosing cohorts that escalate toward the goal of a maximum tolerated dose (MTD) or, for targeted therapies, biologically optimal dose (BOD). Activity/efficacy is not a primary goal. In fact, response rates in phase I trials are seldom more than 10%.2–4 Secondary goals of phase I trials may include scheduling issues, response rate, pharmacokinetic (PK) information (absorption, distribution, metabolism, and elimination [ADME]), and effects on molecular targets or pathways (pharmacodynamics [PD]). These later biologic endpoints are increasingly important components of phase I trials as dose determinants are inherently linked to drug exposure and effect, especially as we move away from more indiscriminant cytotoxic agents and toward the study of molecular-focused therapeutics. These biologic questions are also the basis of comparative oncology modeling of drug development and are emphasized in their design.5,6 Phase I starting dose selection is a critical design question.7 Dose is based on data in normal research on Beagle dogs, extrapolation from rodent data, and even human patient data if such exist.1,3,4 Different options include starting with one-third of the “no observable adverse event level” (NOAEL), or one-tenth of the severe toxicity dose in the most sensitive species, or if normal dog data are available, one-half of the MTD in Beagles as they seem to be less sensitive to adverse events (i.e., toxicity) than are more age-advanced, tumor-bearing patient dogs. Phase I design is integral to the long-term success or failure of an agent’s development. Therefore, if the starting dose is too low, the length of the trials is longer, there is poor use of resources, and the number of patients exposed to suboptimal doses is increased. For targeted therapies, it also makes defining the BOD or therapeutic index more difficult.8,9 As with starting dose, escalation strategies greatly affect the number of patients treated at a potentially ineffective dose, the length of the trial, the use of resources, and the risk of adverse events.7 The traditional method of escalation, outlined in Table 17-2, uses a “3 × 3” cohort design, wherein dose escalations are made with three dogs per dose level and the MTD is set based on the number of patients experiencing a DLT. A DLT is defined as grade III or greater toxicity in any category (except hematologic) according to predefined adverse event categories, such as those in the Veterinary Cooperative Oncology Group—Common Terminology Criteria for Adverse Events (VCOG-CTCAE version 1.1).2,7,10 Grade IV is the cutoff most preferred for myelosuppression DLTs because these events are usually considered manageable with supportive care, generally transient, and often clinically silent.1–4 Additional DLTs are defined on an agent-by-agent basis due to expected toxicities. These can include or exclude some grade III events from being DLTs, again if transient and clinically silent in nature and are prospectively defined in the study protocol. The MTD is defined as the highest dose level in which no more than one of six dogs develops a DLT. Traditionally, a fixed-dose modified Fibonacci method of dose escalation is used, wherein the dose is escalated 100%, 67%, 50%, 40%, and then 33% of the previous dose as the dosing cohorts increase.7,11 Similar to starting at too low of a dose, if the escalations are too conservative, more patients receive a suboptimal dose; conversely, if the escalations are too rapid, more patients are at risk for significant toxicity and the accuracy of the MTD is compromised. Additionally, interdosing cohorts can be added during the study period if more refined escalation or de-escalation is found to be necessary. Table 17-2 Standard Phase I Dose Escalation Scheme MTD, Maximum tolerated dose; DLT, dose-limiting toxicity. From National Cancer Institute: Cancer Therapy Evaluation Program: http://ctep.cancer.gov/protocolDevelopment/templates_applications.htm. Although the phase I MTD approach works well for traditional cytotoxic chemotherapeutics, it may be irrelevant for molecularly targeted drugs, and phase I trials designed to determine the BOD may be more relevant for so-called “static” agents.8,12,13 Trials evaluating the BOD require validated assays that measure target effect in serial tumor samples and/or a surrogate tissue or fluid that documents activity at the molecular level. One example would be measuring γ-H2AX in tumor or surrogate normal tissues after use of a DNA-damaging agent.14,15 This is a marker of double-stranded DNA breaks and has proved effective in both molecularly targeted and traditional agent evaluation. The depiction in Figure 17-1 shows the immunofluorescent measurement of γ-H2AX in canine lymphoma after treatment with topotecan, illustrating its use as a marker of the drug’s biologic effect and PD readout. Classically, studies of inhibition of c-kit phosphorylation in canine mast cell tumors provided the rationale for approval of tyrosine kinase inhibitors (toceranib phosphate and masitinib).16,17 Such pharmacodynamic modulation studies are increasingly important endpoints of phase I and II designs and now more commonly required as proof of mechanism for drug approval. Several good reviews have outlined phase II trial design.4,18–20 The primary goal of phase II trials is, using the MTD or BOD established in phase I, to identify the clinical or biologic activity in defined patient populations (e.g., tumors with a particular histology, tumors with a particular molecular target) and inform the decision to embark on a larger pivotal phase III trial. The traditional phase II design (phase IIA), the single-arm, open-label phase II trial, is a nonrandomized nonblind activity assessment of a novel drug or therapeutic modality that lacks a control group or uses historic controls that are prone to bias (selection, population drift, and stage migration bias).4,18,19 Simplistically, at least 9 to 14 patients with the same histology or molecular target are treated with the investigational drug to test the null hypothesis of insufficient efficacy.21 Assuming the likelihood of spontaneous regression is less than 5% and expecting at least a 25% response rate for the agent to be clinically useful, with a p less than 0.05 (type I [α] error; false positive) and a power of 0.8 (type II [β] error; false negative), if no responses are observed after the initial cases, the study ends. If a response is noted in one of the cases, the accrual is increased to 31 patients to establish a more accurate response rate. If you expect a less robust response rate (e.g., 5% to 20%), the initial accrual number must be increased.22 Sample-size impact on study power calculations will be outlined in a subsequent section. Some have opined that the leading cause of drug failure in later phase development is our overdependence on these unpredictable single-arm, uncontrolled phase II trials in oncology and that, as such, they should be avoided to ensure phase III trial resources are not wasted because of the results of poorly designed phase II trials.23 It used to be considered that the consequence of type I error (false positive) was less deleterious than that of type II error (false negative) because false-positive trials are likely to be repeated, whereas false-negative trials would result in the abandonment of a potentially active treatment.24 The goals of comparative oncology modeling can help minimize type II error by mechanistically defining activity of novel agents through more detailed PK-PD studies. In today’s environment, however, with an abundance of novel drugs to be evaluated, false-positive results are just as serious because they tie up patient and financial resources. With this in mind, the ideal phase II design would be randomized (e.g., between more than one new investigational drug in the pipeline), blinded, and controlled; modifications of this type applied to standard phase II design are discussed subsequently (controlled phase II or phase IIB trials).25 Since the primary goal of phase II trials is assessment of activity/efficacy, endpoints used to evaluate response are critical to the design. With traditional cytotoxic chemotherapeutics, response criteria are fairly straightforward because size or volume is used to assess response according to several published methodologies (e.g., Response Evaluation Criteria in Solid Tumors [RECIST], World Health Organization [WHO]).26–28 These measurements are both repeatable and have criteria that are strictly defining. It is readily evident that such criteria may not be appropriate for newer molecular-targeted agents that are more likely to be cytostatic rather than cytotoxic and result in stabilization of disease rather than in measurable regression.29,30 In such cases, temporal measures such as progression-free survival (PFS) or time to progression (TTP) are appropriate endpoints; however, these often take too long to mature for timely phase II trials.31 Alternatively, an adequate compromise could be progression-free rate (PFR) at predetermined time points. Again, comparative oncology models may more expediently define TTP or PFR due to compressed progression times in our veterinary patients. These measures can also more accurately define response in the minimal residual disease (MRD) setting such as trials interrogating novel adjuvant therapies for canine osteosarcoma (OSA) in the postamputation setting. This will be expanded on in future sections. Secondary endpoints that may be evaluated in phase II trials include QOL assessments, comparative cost of therapy, days of hospitalization, and more detailed adverse event evaluations. Importantly, phase II trials serve to expand our knowledge of the cumulative or long-term toxicities associated with new agents that may not be observed in short-term phase I trials designed to elucidate acute toxicity. An example of this in the veterinary literature involved a combined phase I/II trial simultaneously investigating the safety of liposome-encapsulated doxorubicin (LED) while comparing its activity with native doxorubicin in cats with vaccine-associated sarcomas.32 Unexpectedly, the MTD established for LED in the acute phase I component of the trial was found to result in delayed and dose-limiting nephrotoxicity after long-term follow-up in the phase II component of the trial. Such discoveries are key to defining an agent’s therapeutic window with repeated administrations. New clinical trial concepts have entered into use in great part due to a recent initiative of the Food and Drug Administration (FDA) to allow for “preclinical studies to provide evidence necessary to support the safety of administering new compounds to humans.”33 These are known as phase 0 trials, and they precede the traditional trials defined previously.34,35 The role of phase 0 in cancer drug development is for biomarker and assay development/validation and evaluation of target modulation.33 Phase 0 trials allow for the systematic de-prioritization of investigational agents that exhibit excessive toxicity or fail to show expected biologic effects and are used to direct dose selection for future studies.33 They represent first-in-species trials, usually of a small number of patients, and utilize lower and likely subtherapeutic drug doses. Comparative oncology trials allow the unique opportunity to answer the preclinical questions required within these trials. Phase 0 trials are “proof of concept” studies, and PD effects are measured within target tissues, such as the tumor itself.34,35 These trials can also define surrogate markers of target effect, therapeutic response, or metabolites in surrogate tissues or fluids, such as blood or urine. PK data are also a hallmark of phase 0 trial designs. Biologic endpoints such as PK/PD analysis allow for a much broader understanding of new drug mechanism, therefore informing phase I/II trial design. It has been suggested that if phase II trials are “learning” trials, phase III trials are “confirming” trials.2,19,36 These larger, randomized, blind, and controlled trials have the goal of comparing a new drug or combination with standard-of-care therapies. They are often performed by large co-operative groups, which ensures greater case accrual, and FDA pivotal trials require multiinstitutional involvement. True phase III trials have not been common in veterinary medicine because of their size and expense, but this culture is changing with a broader focus on veterinary oncology drug development and approval. An example of a multicenter phase III trial would be the randomized comparison of liposome-encapsulated cisplatin (SPI-77) versus standard-of-care carboplatin in dogs with appendicular OSA.37 No difference was observed between treatment groups, and SPI-77 did not show an activity advantage, despite allowing five times the MTD of native cisplatin to be delivered in a liposome-encapsulated form.37 This helps define the application of phase III efforts in veterinary oncology. Additionally, there are a number of pivotal veterinary oncology clinical trials that have directly impacted how we treat patients today. Chemotherapy as an adjunct treatment has improved survival in many pets with cancer. This is highlighted in a disease such as canine OSA, in which disease-free survival (DFS) postamputation alone is just 4 months but, with the addition of chemotherapy (cisplatin, carboplatin, or doxorubicin), improves median survival to approximately 1 year.38–42 Although a number of clinical trials have been performed with various schedules and regimens of delivery, all highlight the importance of treatment with chemotherapy in the micrometastatic minimal residual disease setting. These are treatment trials, wherein most studies compare groups to historic controls, but many are prospective in nature to more accurately detect a difference in treatment protocols if one exists. Another definitive example of clinical trials with high clinical impact were those comparing the inclusion of doxorubicin in multiagent chemotherapy protocols for dogs and cats with lymphoma versus COP (cyclophosphamide, vincristine, and prednisone) or single-agent therapy alone.43–48 It was evidenced by a number of protocols that doxorubicin administration contributed significantly to improved survival over those protocols without it, therefore securing its place as the single most efficacious agent against lymphoma.47,48 More recent pivotal trials have included the registration of the first approved veterinary oncology agents, two tyrosine kinase inhibitors (TKIs; Palladia, Pfizer; Kinavet, AB Sciences) for the treatment of mast cell tumors.16,17,49,50 TKIs showed improvement in PFS over placebo controls, and these trials define the process for expanding future efforts in veterinary oncology drug approval. The overriding function of clinical research is to provide a definitive answer to a clinical question. However, it is rarely that simple. It is possible that once complete, clinical trial conclusions may be incorrect based on chance or design error. Chance error results when an erroneous inference is drawn from a study sample group that is not actually representative of an entire patient population. Accounting for this potential error is essential in prospective clinical trial design, and its first critical step is to articulate the study hypothesis.51 Type I or α error (false positive) occurs if an investigator rejects the null hypothesis when it is actually true.36,52 This is also referred to as the study’s level of significance. Type II or β error (false negative) occurs when one fails to reject the null hypothesis when it is actually incorrect.36,52 Type I and II errors are due to chance and cannot be avoided completely, although steps can be taken to reduce their potential impact by increasing sample size and augmenting study design or measurements. Power is the ultimate measure of a clinical trial’s results and also must be prospectively controlled. Power is defined as 1-β, the probability of correctly rejecting the null in the sample if the actual effect in the population is equal to (or greater than) the effect size.36,52 Power is governed by sample size, with the goal being to enroll enough patients to accurately allow for a difference to be seen between groups. Power is irrelevant if the results are statistically significant, but if not, it is important to ensure the study had adequate numbers to detect a difference between groups. If a study to detect the difference between two cancer treatments is designed with an α of 0.05, then the principal investigator (PI) has set 5% as the maximum chance of incorrectly rejecting the null hypothesis if it is true. This is the level of doubt the PI is willing to accept when statistical tests are used to compare the two treatments. If β is set at 0.10, the PI is willing to accept a 10% chance of missing an association of a given effect if it exists. This represents a power of 0.90 (1-β), or a 90% chance of finding an association of that size or greater. α and β levels are determined prior to trial initiation, and their set points are based on the importance of avoiding either a false-positive or a false-negative result. Bias is introduced error in clinical research. One of the main ways to minimize bias is through the use of randomization. Randomization is the process of assigning research participants to a group within a clinical trial by chance instead of choice.2,36,52 Groups include either the investigational group, those to receive the study drug, or the control group, those to receive a placebo (or comparator drug). Each participant enrolling in a trial has an equal chance of receiving the study drug, and the goal is to balance the groups based on participant characteristics (e.g., age, stage of disease, previous treatments) that may influence a response to therapy.2,36,52 At the end of the study, if bias is reduced by randomization, then the result (positive or negative) is more likely to be true. Although this is the ideal way to conduct clinical trials, it is still uncommon in veterinary oncology trials. Unfortunately, in veterinary medicine, historic rather than active controls have been used for comparison of new therapies all too often. It is common in oncology trials to also blind participants so that patients, or in our case clients, do not know which treatment group their pet is in. Blinded or true active comparator trials, although rare in veterinary oncology due in part to limited funding, are becoming more frequent as regulatory registration trials ultimately require them. Reducing bias in clinical trials is an integral concept within trial design. Stratification is a concept used often in human oncology trials and is gaining headway in veterinary oncology trial design as well.36,52,53 This allows for grouping of patients based on known prognostic factors. For example, this might include the stratification of dogs with lymphoma by clinical stage or T- versus B-cell immunophenotype with the goal of creating equal numbers of each within a treatment group. One of the main benefits of stratification is that it can prevent potential bias from known prognostic factors. For example, a treatment cohort may be doing comparatively poorly because the majority of dogs in that cohort had T-cell lymphoma. However, a difficulty with stratification is that the more prognostic factors are controlled, the less power the study has, and thus the need to increase sample size considerably. Randomization to treatment groups should be performed after stratification. Once a drug has been granted a license or registered for a specific label use by the appropriate regulatory body (e.g., the FDA), post-registration phase IV trials may be performed to gain more information on adverse events, long-term risks, off-label benefits, and the economic impact of the agent in the marketplace.54 Essentially, phase IV trials investigate the drug more widely than do the clinical trials used for registration. They may also involve treatment of special populations (e.g., the elderly, pediatric patients, or individuals with renal or hepatic dysfunction).4 The body of data on PK generated from postregistration trials is used to inform decisions on dose in special populations. Productivity and economic differences of one treatment over another, mostly due to insurance reimbursement requirements on the human side, are also a consideration in data collection and evaluation in phase IV clinical trials. When designing clinical trials it is key to define provisions that create uniformity and consistency in clinical research. Their primary intent is to ensure the safety of trial materials and participants and the integrity of clinical data. Such provisions determine good manufacturing practice (GMP) and good clinical practice (GCP). GMP principles ensure the safe manufacture and testing of pharmaceutical drugs, biologics, and medical devices by outlining aspects of production that can impact the quality of a product. GCP guidelines were devised by the International Conference on Harmonization (ICH) to protect the rights and safety of human patients in clinical trials.36,55 GCP includes standards on how clinical trials should be designed and conducted, defines the roles and responsibilities of trial sponsors and clinical research investigators, and monitors the reporting of trial data.55 It requires the use of a standardized clinical protocol and strict documentation of procedures and adherence to that protocol. GCP also establishes guidelines for oversight by external bodies, such as the Institutional Review Board (IRB) and the Data Safety and Monitoring Board (DSMB).55 The IRB serves as an independent ethics committee that has the power to approve, disapprove, or ask for modifications to planned study protocols. IRBs traditionally were institutional entities involved with in-house clinical trial oversight but now have grown in scope and purpose. IRBs must include at least one nonscientist and a noninstitutional member known as a community member. If a clinical trial includes a vulnerable population, then the IRB must include an expert in the needs and specialties of this group. The IRB reviews protocol ethics, informed consent, and possible conflicts of interest and provides scientific review of study protocol and results.36,55 IRB review and approval occur both prior to study initiation and periodically within the conduct of a clinical trial to ensure ethical standards are being met throughout the trial process. The DSMB (sometimes called a Data Monitoring Committee) is a third-party panel of experts consisting of at least one statistician and is populated by clinicians expert in the field of research or drug of study.36,56 In long-term and high-impact studies an ethicist and/or a patient advocate representative may also serve on the DSMB. The purpose of the DSMB is to ensure the safety of participants, the validity of data, and the appropriate termination of studies for which significant benefits or risks have been uncovered or if it appears that the trial cannot be concluded successfully. The main role of the DSMB is to oversee serious adverse events (SAE) and their management to ensure patient safety.56 However, the DSMB also reviews interim study results to determine if an overwhelming benefit is evident in either the study or the control arm or if it is unlikely the study will answer the proposed study aim. The latter results if interim data reveal continuance is unlikely to produce a statistically significant result, and the study will then be terminated prematurely. It is important for all safety monitoring to be contemporaneous, including disclosure to the FDA of SAEs in real time. GCP compliance is necessary for FDA registration trials in both veterinary and human oncology drug development. Oversight of GMP and GCP provisions are provided by the FDA (http://www.fda.gov), and in the European Union (EU), oversight is provided by the European Medicines Agency (EMA; http://www.ema.europa.eu).
Clinical Trials and Developmental Therapeutics

Phase I Trials (Dose Finding)

Phase II Trials (Activity/Efficacy Trials)
Endpoints of Activity/Efficacy
Phase III Trials (Pivotal/Confirming Trials)
Sample Size and Power
Randomization
Phase IV Trials (Postregistration Trials)
Good Manufacturing Practice/Good Clinical Practice Criteria
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Veterian Key
Fastest Veterinary Medicine Insight Engine