Chapter 6 CANINE HYPERADRENOCORTICISM (CUSHING’S SYNDROME)
In 1932 Dr. Harvey Cushing described eight humans with a disorder that he suggested was “the result of pituitary-basophilism.” Six of those eight patients had small, basophilic pituitary adenomas and clinical features of excess adrenocortical cortisol secretion. Careful study of these and other individuals diagnosed years ago suggests numerous primary causes for what was then considered a single condition. Chronic excesses in the serum cortisol concentration, however, represent the final common denominator of their illnesses. The eponym Cushing’s syndrome is an “umbrella term” referring to the constellation of clinical and chemical abnormalities that result from chronic exposure to excessive concentrations of glucocorticoids. The eponym Cushing’s disease is applied to cases of Cushing’s syndrome in which hypercortisolism occurs secondary to inappropriate excessive secretion of adrenocorticotropic hormone (corticotropin, ACTH) by the pituitary; that is, pituitary-dependent hyperadrenocorticism (PDH). Canine hyperadrenocorticism (canine Cushing’s syndrome [CCS]) also has various pathophysiologic origins, but all have that common denominator: chronic excesses in systemic cortisol.
REGULATION OF GLUCOCORTICOID SECRETION
Corticotropin-Releasing Hormone (CRH)
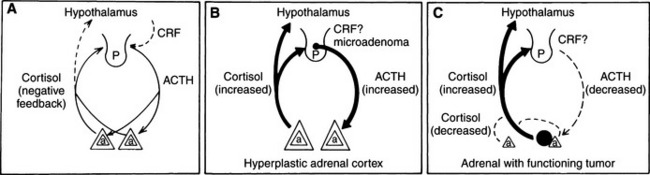
With an understanding of the portal circulation, which “connects” the hypothalamus and the pituitary, came an appreciation that the hypothalamus exerts control over secretion of ACTH by the anterior pituitary. ACTH, in turn, exerts control over adrenocortical secretion of cortisol. Cortisol, in part, then completes the circle by inhibiting secretion of hypothalamic and pituitary hormones (Fig. 6-1). The factor released by the hypothalamus is CRH, a polypeptide containing 41 amino acid residues. The CRH-secreting neurons are located in the anterior portion of the paraventricular nuclei in the hypothalamus. CRH has a long plasma half-life (approximately 60 minutes). Both arginine vasopressin and angiotensin II potentiate CRH secretion and, in turn, ACTH in humans. In contrast, oxytocin inhibits CRH-mediated ACTH secretion in humans. Regulatory roles in the physiology of ACTH secretion for arginine vasopressin, oxytocin, and angiotensin II have not been consistently demonstrated in dogs, but the importance of CRH in controlling normal ACTH release has been supported (Kemppainen and Sartin, 1987; Kemppainen et al, 1992).

Adrenocorticotropic Hormone (ACTH)
ACTH is a 39-amino acid peptide hormone (MW 4500) processed from a large precursor molecule, pro- opiomelanocortin (POMC) (MW 28, 500). In the pituitary cells responsible for the synthesis of ACTH (corticotrophs), messenger ribonucleic acid (mRNA) directs the synthesis of POMC and its processing into smaller, biologically active fragments (Fig. 6-2). The function and importance of these peptide fragments—β-lipotropin (β-LPH), α–melanocyte-stimulating hormone (α-MSH), β-MSH, β-endorphin, and N-terminal fragment—constitute an evolving area of endocrine study. Most of these peptides are glycosylated to some extent, resulting in their various reported molecular weights. The basophilic staining characteristics of corticotrophs can be explained by the carbohydrate nature of these moieties (Aron et al, 2001a).
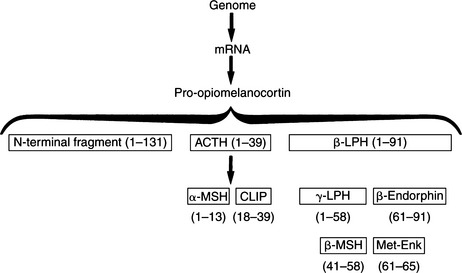
Two of the fragments depicted in Fig. 6-2 are contained within the structure of ACTH and therefore are byproducts of ACTH metabolism: α-MSH is identical to ACTH1-13, and corticotropin-like intermediate-lobe peptide (CLIP) is identical to ACTH18-39. Neither of these peptides is secreted as a separate hormone in humans. β-Endorphin may act as an “endogenous opiate,” which suggests a role in pain sensation. It may also affect endocrine regulation of other pituitary hormones and may have a role in neural control of breathing (Aron et al, 2001a). Plasma concentrations of the N-terminal fragment have been demonstrated to increase in response to hypoglycemic stress; this fragment may also be an adrenal growth factor and/or may potentiate ACTH action on steroidogenesis. The physiologic function of β-LPH is not well understood. However, it is known that both β-LPH and β-endorphin have the same secretory dynamics as ACTH; that is, they increase in response to stress or hypoglycemia and are suppressed by glucocorticoids. These hormones also parallel ACTH in disease states. For example, concentrations increase in Addison’s disease, pituitary Cushing’s disease, and “Nelson’s syndrome” (growing pituitary tumor after removal of the adrenals) (Peterson et al, 1986). Concentrations of β-LPH and β-endorphin decrease in dogs with autonomously secreting adrenocortical tumors.
Both CRH and ACTH are secreted in a pulsatile manner, with a diurnal rhythm in humans that results in a peak before awakening followed by a progressive decline in concentrations throughout the day. Diurnal rhythms have not been established in dogs (Kemppainen and Sartin, 1984 and 1987; Murase et al, 1988). ACTH secretion also increases in response to feeding in both humans and animals (Aron et al, 2001a). The primary function of ACTH is to stimulate the secretion of glucocorticoids from the adrenal cortex. The stimulatory properties of ACTH on adrenocortical mineralocorticoid secretion and on androgenic steroids are less important. The amino terminal end of the ACTH molecule (amino acids 1 to 18) is responsible for its biologic activity.
Many types of stress stimulate secretion of ACTH, often superseding normal daily fluctuations. Physical, emotional, and chemical stressors such as pain, trauma, hypoxia, acute hypoglycemia, cold exposure, surgery, and pyrogens have been demonstrated to stimulate ACTH and cortisol secretion (Aron et al, 2001a). The increase in ACTH concentrations during stress is mediated by vasopressin and CRH. Although physiologic cortisol levels do not blunt the ACTH response to stress, high doses of exogenous corticosteroids do suppress ACTH.
Negative feedback by cortisol and synthetic glucocorticoids on ACTH secretion occurs at both the hypothalamic and pituitary levels and appears to act by two mechanisms: “Fast feedback” is sensitive to the rate of change in the cortisol concentration, whereas “slow feedback” is sensitive to the absolute cortisol concentration. The first mechanism is probably nonnuclear; that is, this phenomenon occurs too rapidly to be explained by the influence of corticosteroids on nuclear transcription of specific mRNA responsible for ACTH. Slow feedback, which occurs later, may be explained by a nuclear-mediated mechanism and a subsequent decrease in the synthesis of ACTH. This latter form of negative feedback is the type probed by the dexamethasone suppression test. In addition to the negative feedback by adrenal steroid secretion, ACTH also exerts a negative feedback effect on (i.e., inhibits) its own secretion (short loop feedback), as depicted in Fig. 6-1 (Aron et al, 2001a).
Steroids
ZONES OF THE ADRENAL CORTEX AND THEIR PRODUCTS.
The major hormones secreted by the adrenal cortex are cortisol and aldosterone. Histologically, the adrenal cortex is composed of three zones. The scheme of adrenal steroidogenic synthesis has been clarified by analysis of the enzymes involved in the synthesis of these hormones. Most of the enzymes belong to the family of cytochrome P450 oxygenases (Table 6-1). Because of the enzymatic differences between the zona glomerulosa and the inner two zones, the adrenal cortex functions as two separate units, with differing regulation and secretory products.

The outer zona glomerulosa produces aldosterone and is deficient in 17α-hydroxylase activity (new enzyme name, CYP17), which renders this zone incapable of synthesizing cortisol or androgens. In contrast, only cells in the zona glomerulosa contain the enzymes necessary for dehydrogenation of 18-hydroxycorticosterone to synthesize aldosterone. Aldosterone synthesis is regulated primarily by the renin-angiotensin system and by serum potassium concentrations (see Fig. 6-1, B and Fig. 6-3). The middle zona fasciculata, the thickest of the three adrenocortical layers, functions as a unit with the narrow, inner zona reticularis; these are the two zones from which cortisol and androgens are produced. Only cells in these two layers of the adrenal cortex have 17α-hydroxylase (CYP17) activity and can synthesize 17α-hydroxypregnenolone and 17α-hydroxyprogesterone, precursors of cortisol and adrenal androgens (Fig. 6-4). These zones are regulated primarily by ACTH. The conversion of cholesterol to pregnenolone is the rate-limiting step in adrenal steroidogenesis and the major site of ACTH action, via activation of adenylate cyclase, which increases cyclic adenosine monophosphatase (AMP) and then activation of intracellular phosphoprotein kinases (Fig. 6-5) (Aron et al, 2001b).
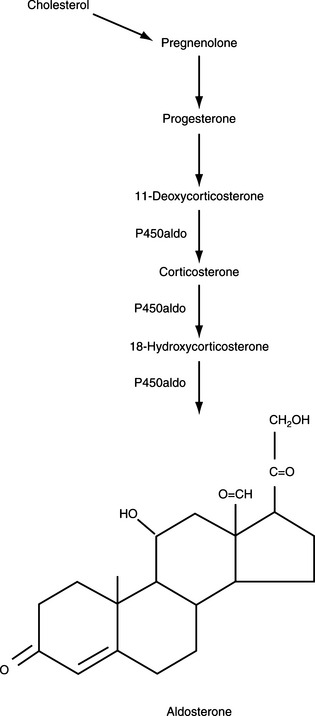
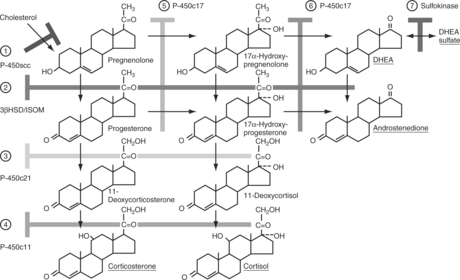
FIGURE 6-4 Steroid biosynthetic pathway in the adrenal cortex. The branching pathways for glucocorticoids, mineralocorticoids, and adrenal androgens and the structures of these steroids and their biosynthetic precursors are shown. The names of the biosynthetic enzymes are shown in the boxes. See Table 6-1 for the nomenclature of steroidogenic enzymes.
STEROIDOGENESIS.
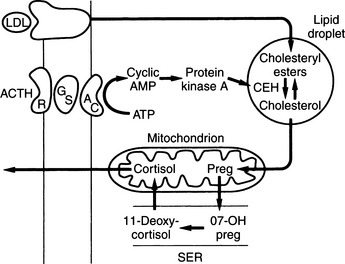
Cortisol, aldosterone, androgens, and estrogens are “steroids.” Synthesis of these steroids begins from enzymatic action on cholesterol (see Figs. 6-3 and 6-4). Plasma lipoproteins are the major source of adrenal cholesterol, though synthesis in the gland from acetate also occurs. Low-density lipoprotein (LDL) accounts for about 80% of cholesterol delivered to the adrenals, and a small pool of free cholesterol is available in the adrenals for rapid response to stimulation. When stimulation occurs, there is also increased hydrolysis of stored cholesteryl esters to free cholesterol, increased uptake from plasma lipoproteins, and increased cholesterol synthesis in the adrenals. The acute response to a steroidogenic stimulus is mediated by the steroidogenic acute regulatory (StAR) protein. This mitochondrial phosphoprotein enhances cholesterol transport from the outer to the inner mitochondrial membrane (Aron et al, 2001b).
The conversion of cholesterol to pregnenolone is the rate-limiting step in adrenal steroidogenesis and the major site of ACTH action on the adrenal. Conversion to pregnenolone occurs in the mitochondria and involves two hydroxylations, followed by the side-chain cleavage of cholesterol (see Fig. 6-5). A single enzyme, CYP11A1, mediates this process; each step requires molecular oxygen and a pair of electrons. After synthesis, pregnenolone is transported out of the mitochondria. Cortisol is the focus of this chapter, and its synthesis proceeds via 17α-hydroxylation in smooth endoplasmic reticulum (Aron et al, 2001b). By means of the steps outlined in Fig. 6-4, cortisol is continuously synthesized, and under basal conditions in the dog, this typically results in serum concentrations that range from 1 to 5 μg/dl.
REGULATION OF SECRETION.
ACTH is the trophic hormone of the zonae fasciculata and reticularis. Delivery of ACTH to the adrenal cortex leads to rapid synthesis and secretion of cortisol and androgens. Secretion of ACTH, in turn, is regulated by the hypothalamus and central nervous system via neurotransmitters, CRH, and arginine vasopressin. The plasma concentration of cortisol increases within minutes of ACTH administration. ACTH increases RNA, deoxyribonucleic (DNA), and protein synthesis. Chronic stimulation leads to adrenocortical hyperplasia and hypertrophy; conversely, ACTH deficiency results in decreased steroidogenesis and is accompanied by adrenocortical atrophy, decreased weight of the gland, and decreased protein and nucleic acid content (Aron et al, 2001b).
PATHOPHYSIOLOGY
Pituitary-Dependent Hyperadrenocorticism
PATHOLOGY IN HUMANS.
“ACTH-secreting pituitary tumors exist in virtually all patients with (pituitary) Cushing’s disease” (Aron et al, 2001a and 2001b). These tumors are usually benign adenomas less than 10 mm in diameter. About 50% of these tumors are 5 mm or smaller in diameter, and microadenomas as small as 1 mm have been described. The tumors identified in people with Cushing’s disease are either basophilic or chromophobe adenomas and may be found anywhere in the anterior pituitary. About 15% to 25% of ACTH-secreting tumors are large and have invasive tendencies. Malignant tumors have been reported. Diffuse hyperplasia of anterior pituitary corticotrophs, or adenomatous hyperplasia, which is presumed to result from excess CRH, is poorly defined and occurs rarely. The adrenal glands in people with Cushing’s disease are thickened due to hyperplasia of both the zona fasciculata and the zona reticularis; the zona glomerulosa is normal. In some cases, ACTH-secreting pituitary adenomas cause bilateral nodular hyperplasia. These adrenals are diffusely hyperplastic and have one or more nodules that vary in size from microscopic to several centimeters in diameter, although the most common presentation is multiple small nodules (Aron et al, 2001b).
PITUITARY CONTROL AND FEEDBACK.
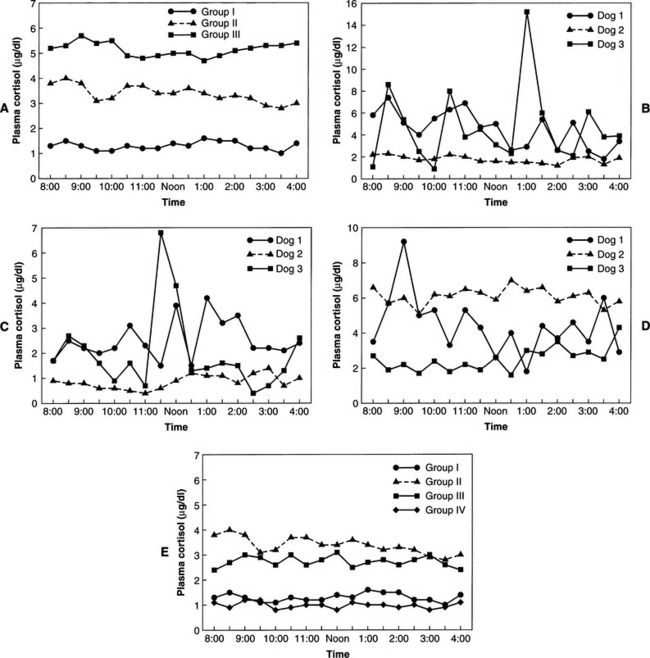
In normal individuals (humans and animals), ACTH secretion appears to be both random and episodic. This appearance is misleading, because although ACTH is secreted episodically, it functions exquisitely well in maintaining plasma cortisol concentrations at the levels required for homeostasis. Secretion, therefore, is not “random.” The most common abnormality in pituitary-dependent hyperadrenocorticism (PDH) is the fact that both the frequency and amplitude of ACTH secretory “bursts” are chronically excessive. Chronic excesses in ACTH secretion result in excess cortisol secretion and, eventually, adrenocortical hyperplasia. Feedback inhibition of ACTH secreted from a pituitary adenoma by physiologic or excess levels of glucocorticoids is relatively ineffective (Fig. 6-6). If glucocorticoids were “normally” effective in negative feedback inhibition of ACTH secretion, PDH would not evolve. In dogs with naturally occurring Cushing’s disease, episodic secretion of ACTH and, in turn, cortisol results in fluctuating plasma concentrations of both hormones that are often within the normal or reference ranges for most laboratories (Fig. 6-7).
“NORMAL” CORTISOL CONCENTRATIONS IN HYPERADRENOCORTICISM.
Studies of cortisol production in dogs or people with hyperadrenocorticism, such as urine cortisol excretion over 24 hours, easily illustrate the existence of excessive cortisol secretion. The combination of excessive secretion and absence of diurnal variation (if it exists in dogs) in glucocorticoid secretion causes the clinical manifestations of Cushing’s syndrome. The excessive secretion of cortisol is not readily appreciated by randomly assaying basal cortisol concentrations. As shown in Fig. 6-7, most dogs with hyperadrenocorticism usually have plasma cortisol concentrations within the reference or “normal” range at any given moment. The excess cortisol becomes readily apparent when one evaluates the “area under a plasma cortisol curve” over time for dogs with Cushing’s syndrome, especially when those findings are compared with data from healthy dogs. The dog or cat with hyperadrenocorticism is exposed to more cortisol, on a daily basis, than is a normal animal. This chronic and persistent abnormality results in the clinical syndrome associated with cortisol excess.
LOSS OF HYPOTHALAMIC CONTROL.
One reflection of excessive ACTH secretion is the absence of stress responsiveness. Stimuli such as hypoglycemia or surgery fail to further stimulate ACTH and cortisol secretion. Chronic hyperadrenocorticism suppresses hypothalamic function and CRH secretion. Hypothalamic control of ACTH secretion is lost (Aron et al, 2001a), probably owing to suppression of hypothalamic function and CRH secretion as a result of chronic hypercortisolism. Thus chronic hypercortisolism causes a loss of hypothalamic control of ACTH secretion (Aron et al, 2001b; van Wijk et al, 1992).
INCIDENCE OF PITUITARY TUMORS.
Eighty percent to 85% of dogs with naturally occurring Cushing’s syndrome have PDH; that is, excessive secretion of ACTH by the pituitary, causing bilateral adrenal hyperplasia and excessive secretion of glucocorticoids (Feldman, 1983a-c,). The reported incidence of recognized pituitary tumors in dogs with PDH varies tremendously but likely depends on the competence and persistence of the pathologist, as well as the microdissection capabilities and staining capacities of the laboratory performing the histologic examination. It has been reported that more than 90% of dogs with PDH have a pituitary tumor (Zerbe, 1992). Our experience is similar; virtually 100% of dogs with naturally occurring pituitary-dependent Cushing’s syndrome have a pituitary tumor.
PARS DISTALIS VERSUS PARS INTERMEDIA.
In humans and dogs, the pituitary gland is distinctly divided into an anterior section (pars distalis) and a posterior section (pars nervosa). However, dogs, unlike humans, also have a distinct area that separates the anterior and posterior lobes of the pituitary, the pars intermedia (Peterson et al, 1982a). Furthermore, the pars intermedia has been shown to have two distinct cell types (Halmi et al, 1981; Jackson et al, 1981). The predominant cells (A cells) immunostain intensely for α-MSH but only weakly for ACTH. The second population of pars intermedia cells (B cells) stain strongly for ACTH and only weakly for α-MSH. The intense ACTH staining of pars intermedia B cells is similar to the staining characteristics of ACTH-producing pars distalis cells (Peterson et al, 1986).
Pars distalis POMC (the ACTH prohormone molecule; see Fig. 6-2) and, therefore, ACTH secretion are regulated primarily by the interaction of the stimulatory hypothalamic peptide (CRH) and the inhibitory adrenocortical glucocorticoids. The pars intermedia, however, is under negative regulation by dopamine secreted from the arcuate nucleus, as well as by serotonin and CRH. Thus the pars distalis is devoid of a nerve supply and is controlled by hypothalamic CRH that reaches it through the hypophyseal portal vessels, whereas the relatively avascular pars intermedia is innervated and controlled by dopaminergic and serotoninergic fibers from the brain (Peterson et al, 1982a; Peterson, 1984).
Almost all dogs with PDH have a pituitary adenoma, most derived from the pars distalis. However, some dogs with hyperadrenocorticism have been diagnosed with “A cell” pars intermedia adenomas and others with “B cell” pars intermedia adenomas. With extreme rarity, hyperadrenocorticism has been claimed to be caused by pituitary hyperplasia. Even more confusing are individual dogs with (1) two pituitary adenomas, each tumor apparently arising from a different pituitary lobe, or (2) both a tumor and hyperplasia of the pituitary. A small number of dogs have been described as having a functioning pituitary carcinoma. As is quickly appreciated, pituitary “hyperadrenocorticism” is a syndrome with the potential for multiple causes (Peterson et al, 1986; Peterson, 1987). The final common pathway for these disorders remains similar, however. Chronic systemic cortisol excess due to adrenocortical hyperplasia results from chronic and excessive secretion of pituitary ACTH. Clinically, the syndrome of cortisol excess is similar regardless of the underlying pathogenesis or the source of the cortisol.
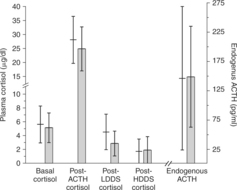
Investigators have attempted to distinguish dogs with pars distalis tumors from those with pars intermedia A-cell or B-cell tumors. It has been suggested that dogs with pars distalis tumors and those with B-cell pars intermedia tumors have excess endogenous ACTH and that their plasma cortisol concentrations are suppressible with large doses of glucocorticoids. The A-cell pars intermedia tumors may not be suppressible (Peterson et al, 1986; Peterson, 1987). Our experience has been different; we have not found it possible to easily distinguish the cause based on antemortem testing (Fig. 6-8) (Leroy and Feldman, 1989). Furthermore, such antemortem testing is likely affected by numerous other factors (e.g., age, breed, duration of illness, tumor size, and benign or malignant nature of the tumor). These discussions currently are of academic interest and have not yet been demonstrated to have clinical significance.
ETIOLOGY OF PDH.
Both a primary pituitary abnormality (ACTH-secreting adenoma) (Aron et al, 2001b) and a central nervous system (CNS) derangement with excessive stimulation of pituitary corticotrophs by CRH or other hypothalamic factors (Krieger, 1983) have been proposed in the past. It was suggested that chronic stimulation of pituitary corticotrophs by hypothalamic CRH could lead to excessive secretion of ACTH, pituitary “hyperplasia,” and eventually neoplastic transformation of some corticotrophs, resulting in a “polyclonal” tumor. However, CRH concentrations in the cerebrospinal fluid of dogs with PDH have been demonstrated to be decreased, whereas ACTH concentrations were normal, despite the syndrome of excess cortisol secretion (van Wijk et al, 1992). Adenomas of the pars distalis are the most common histologic finding in canine PDH, and they represent the best evidence that pituitary tumors are a primary and autonomous cause of the disorder.
We also believe that humans with Cushing’s syndrome represent an excellent and accurate model of the condition in dogs. Virtually all peer-reviewed evidence indicates that humans with pituitary-dependent Cushing’s syndrome have a primary pituitary disorder and that hypothalamic abnormalities (when present) arise secondary to cortisol excess. The endocrine abnormalities in human pituitary-dependent Cushing’s syndrome are (1) excess secretion of ACTH, with bilateral adrenocortical hyperplasia and hypercortisolemia; (2) absent circadian periodicity of ACTH and cortisol secretion; (3) absent responsiveness of ACTH and cortisol to stress (e.g., hypoglycemia or surgery); (4) abnormal negative feedback of ACTH secretion by glucocorticoids; and (5) subnormal responsiveness of growth hormone (GH), thyroid-stimulating hormone (TSH), and gonadotrophs to stimulation (Aron et al, 2001a).
Further evidence that pituitary Cushing’s syndrome is a primary pituitary disorder is based on the high frequency of pituitary adenomas, the response to their removal (in people and dogs), and the interpretation of hypothalamic abnormalities as occurring secondary to hypercortisolemia. People and dogs in which pituitary tumors have been removed, with resolution of their condition, virtually never have a recurrence. Recurrences should be common if the underlying problem were hypothalamic, because regrowth of the tumors should occur. In addition, molecular studies have found that nearly all corticotroph adenomas are monoclonal (Gicquel et al, 1992; Biller, 1994; Faglia, 1994; Orth, 1995). These findings suggest that ACTH hypersecretion arises from a naturally developing pituitary adenoma and that the resulting hypercortisolism suppresses the normal hypothalamic-pituitary axis and CRH release, thereby abolishing hypothalamic regulation of ACTH and cortisol (Aron et al, 2001a).
Other support for the theory that pituitary Cushing’s syndrome is a primary disorder in dogs can be traced to the results of various therapeutic trials that used agents directed at a hypothalamic disorder. In dogs, cyproheptadine, an antiserotonin agent, was therapeutically effective in only three of 24 dogs with PDH, and only one of the three was considered a complete treatment success (Drucker and Peterson, 1980; Stolp et al, 1984). Bromocriptine, a dopamine agonist, was successful in only one of seven dogs with PDH (Peterson, 1987). The dopamine agonists l-deprenyl and pergolide have failed to demonstrate efficacy in the treatment of dogs with pituitary Cushing’s syndrome (Reusch et al, 1999; Tobin et al, 1998). Therefore the concept that such medications are safe and potentially effective, as has been suggested (Bruyette et al, 1997; Peterson, 1999), must be seriously questioned. It is safe to propose that PDH may result from several different physiopathogenic mechanisms but that a hypothalamic cause has not yet received strong scientific support.
EFFECT OF GLUCOCORTICOID EXCESS ON PITUITARY FUNCTION.
In addition to its systemic effects, an excess of glucocorticoids also inhibits normal pitu-itary and hypothalamic function, including the release of TSH, GH, and gonadotropin (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]). Inhibition of secretion of these trophic hormones results in reversible secondary hypothyroidism (TSH), failure to cycle in females or testicular atrophy in males (FSH and LH), and failure to grow in puppies (GH), as well as short stature in children.
Adrenal Tumor Hyperadrenocorticism (ATH)
Primary adrenocortical tumors, both adenomas and carcinomas, apparently develop autonomously. Functioning adrenocortical tumors secrete excessive amounts of cortisol independent of pituitary control. Thus the steroid products of these tumors suppress hypothalamic CRH and circulating plasma ACTH concentrations. Remaining POMC peptides (except α-MSH) are also suppressed by the negative feedback effects of the autonomously secreted glucocorticoids (Peterson et al, 1986). The result of this chronic negative feedback is cortical atrophy of the uninvolved adrenal and atrophy of all normal cells in the involved adrenal (see Fig. 6-6).
Little is known about the events leading to formation of adrenocortical tumors. Molecular defects, including activating mutations of receptors for corticotrophic factors, have been suspected as contributing to this process. Structural mutations of the corticotropin-receptor gene have not been detected in adrenocortical tumors (Reicke et al, 1997). However, some of these tumors have had gastric inhibitory polypeptide receptors, vasopressin receptors, receptors for β-adrenergic response and, more recently, receptors for immune cells and their cytokine products (Lacroix et al, 1997; Willenberg et al, 1998). For example, high local concentrations of interleukin-1 combined with aberrant expression of type I interleukin-1 receptors by a particular population of adrenocortical cells could have been followed by clonal expansion of those cells, thus providing a basis for tumor formation.
Cortisol secretion by adrenocortical tumors is episodic and random (see Fig. 6-7). However, most if not all of these tumors appear to retain ACTH receptors, because they respond to exogenous administration of ACTH by synthesizing and secreting cortisol. These adrenocortical tumors are typically unresponsive to manipulation of the hypothalamic-pituitary axis with pharmacologic agents such as dexamethasone. There have been no consistent clinical or biochemical features that aid in distinguishing dogs or cats with functioning adrenal adenomas from those with adrenal carcinomas. The only characteristic considered somewhat consistent is that adrenocortical carcinomas tend to be larger than adenomas (Reusch and Feldman, 1991).
Ectopic ACTH Syndrome
Ectopic ACTH syndrome has not yet been diagnosed in the dog. In humans it comprises a varying group of tumors that are capable of synthesizing and secreting ACTH, which in turn ultimately causes adrenocortical hyperplasia and hypercortisolism. Tumors with the potential for causing ectopic ACTH syndrome in humans include oat cell (small cell) carcinoma of the lung, thymoma, pancreatic islet cell tumors, carcinoid tumors (lung, gut, pancreas, and ovary), medullary carcinoma of the thyroid, pheochromocytoma, and pulmonary tumorlets (Arioglu et al, 1998; Aron et al, 2001b). When such tumors synthesize and secrete excessive amounts of biologically active ACTH, related β-LPH and β-endorphin peptides are also synthesized and secreted in excess, as are inactive ACTH fragments. CRH-like activity has also been demonstrated in ectopic tumors that secrete ACTH; however, secretion of CRH into plasma has not been demonstrated, and the role of tumor-derived CRH is unclear (Tyrrell et al, 1991; Bertagna, 1994; Becker and Aron, 1994).
Adrenocortical Nodular Hyperplasia
Macronodular hyperplasia of the adrenals occurs in about 20% of people with adrenocortical hyperplasia. Virtually all of these people are thought to have pituitary-dependent Cushing’s disease. One or both adrenals are usually grossly enlarged and have multiple nodules of varying size in the cortex (Bertagna, 1992). Dogs and cats with bilateral adrenocortical nodular hyperplasia are also well recognized, accounting for 5% to 10% of hyperadrenocorticism cases. The exact pathogenesis of this syndrome is unclear, although, as in humans, virtually all these dogs and cats are presumed to represent an anatomic variant of PDH.
Rarely, humans have ACTH-independent bilateral nodular adrenocortical hyperplasia (Malchoff et al, 1989; Samuels and Loriaux, 1994). It is conceivable that some non-ACTH factor stimulates abnormal adrenocortical cells. On investigation, several humans with one subset of this syndrome had clinical features of hyperadrenocorticism, adrenocortical nodular hyperplasia, subnormal morning plasma cortisol concentrations, and suppressed responsiveness to exogenous ACTH (Lacroix et al, 1992; Reznik et al, 1992). Food intake stimulated cortisol secretion in these people (Hamet et al, 1987). Each showed inappropriate adrenocortical sensitivity to normal postprandial increases in the secretion of gastric inhibitory polypeptide (GIP). In view of the poor homology between GIP and ACTH, it was unlikely that the adrenocortical ACTH receptors were modified to bind GIP (Bertagna, 1992).
Unilateral versus Bilateral Adrenal Tumors
BILATERAL ADRENOCORTICAL TUMORS.
Hyperadrenocorticism caused by bilateral adrenocortical neoplasia is rare in dogs. In four such dogs, three had bilateral adrenocortical adenomas and one had bilateral carcinomas. In a study of 15 dogs with Cushing’s syndrome caused by functioning adrenocortical tumors, three (20%) had bilateral tumors (Hoerauf and Reusch, 1999). However, we believe that the incidence of bilateral adrenocortical tumors is far lower than is implied in the latter study. In such cases, the history, physical examination findings, clinicopathologic abnormalities, and results of ACTH stimulation and low-dose dexamethasone suppression tests should be compatible with a diagnosis of hyperadrenocorticism. Adrenocortical neoplasia can usually be differentiated from PDH by finding lack of plasma cortisol suppression after administration of a high dose of dexamethasone, undetectable endogenous ACTH concentrations, and identification of the adrenal masses on abdominal ultrasonography (Ford et al, 1993; Hoerauf and Reusch, 1999). A review of this condition is presented in the ultrasonography section of this chapter.
ADRENOCORTICAL TUMOR AND PHEOCHROMOCYTOMA.
We have diagnosed several dogs with a pheochromocytoma (adrenal medullary tumor) in one adrenal and an adrenocortical tumor in the contralateral gland. This can be confusing, because ultrasonography may reveal bilateral adrenomegaly, and endocrine testing suggests an adrenocortical tumor (Von Dehn et al, 1995). This is a relatively uncommon, but not rare, condition. As adrenal ultrasonography continues to expand its role as a frequent and valuable diagnostic aid, these diagnoses likely will become more frequent. A review of this condition is presented in the ultrasonography section of this chapter.
Simultaneous Pituitary Tumor and Adrenal Cushing’s Syndrome
Several dogs with both a functioning adrenocortical tumor and a pituitary microadenoma or macroadenoma (and bilateral adrenocortical hyperplasia) have been reported (Greco et al, 1999). Endocrine evaluation of such dogs has been diagnostic for hyperadrenocorticism. However, tests to distinguish between pituitary-dependent and adrenocortical tumor hyperadrenocorticism provide confusing or contradictory results. This must be considered an extremely rare condition. A review of this syndrome and its diagnosis and management is presented in the diagnosis section of this chapter.
PATHOLOGY
Pituitary
MICROADENOMAS.
Eighty percent to 85% of dogs with naturally developing hyperadrenocorticism have pituitary-dependent disease. The reported incidence of histologically recognized pituitary tumors varies between 20% and 100% (Meijer, 1980; McNicol, 1987). Pituitary tumors in dogs, like those in humans, can be smaller than 1 mm in diameter. At the time of diagnosis, it is well documented that approximately 50% of dogs with pituitary Cushing’s disease have tumors less than 3 mm in diameter. The remainder of evaluated dogs with PDH, specifically those without CNS signs, had tumors 3 to 12 mm in diameter (Bertoy et al, 1995). Recognition of some pituitary tumors requires careful microdissection, experience, special stains, and a great deal of patience. Because these criteria are not always met, the reported incidence of recognized pituitary tumors in dogs with PDH is underrepresented. Tumors larger than 3 mm in diameter should be grossly visible, easier to recognize, and more likely to be identified than the smaller masses.
Most ACTH-secreting pituitary tumors are defined as microadenomas because they are less than 1 cm in diameter (Aron et al, 2001a). They are not usually encapsulated but may be surrounded by a rim of compressed normal pituitary cells. With routine histologic stains, these tumors are typically composed of compact sheets of well-granulated basophilic cells in a sinusoidal arrangement. ACTH-secreting adenomas show Crooke’s changes (a zone of perinuclear hyalinization that results from chronic exposure of corticotrope cells to hypercortisolism). Electron microscopy demonstrates secretory granules that vary in size from 200 to 700 nm. The number of granules varies in individual cells.
MACROADENOMAS.
A significant percentage of dogs with PDH (at least 10% and more likely more than 20%) have large pituitary tumors (Duesberg et al, 1995). A macroadenoma is defined as a tumor visible on gross examination of the pituitary, or greater than 1 cm in diameter. These tumors have the potential to compress or invade adjacent structures as they expand dorsally beyond the confines of the sella turcica. The masses usually extend dorsally into the hypothalamus, often causing signs (see page 296). Because the canine sella turcica is shaped like a saucer rather than a cup (as in humans), bony destruction of the sella walls has not been observed in dogs with expanding tumors. Large, expanding masses need not contact bone to expand into the overlying structures of the brain. Such tumors may appear chromophobic on routine histologic study, but they typically contain ACTH and its related peptides. Malignant pituitary tumors occur but are uncommon.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


