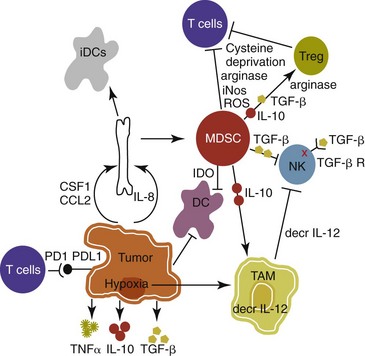
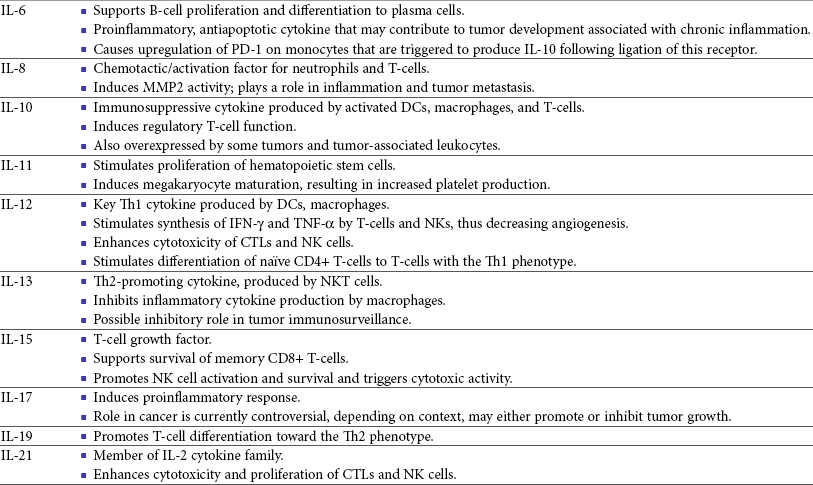
13 With the advancement and sophistication of techniques used to study the immune system comes the increased ability to precisely target the malignant cells while leaving normal tissues intact. One of the major goals of immunotherapy is to develop a potent, long-lasting antitumor immune response without producing untoward side effects. However, it is difficult to develop an immunotherapy that is not affected by tumor-induced immune tolerance or even the immune system itself. For example, therapy with the T-cell growth factor cytokine, interleukin-2 (IL-2), leads to expansion of regulatory T-cells, known for their powerful immunosuppressive abilities.1 In addition, when considering monoclonal antibody (MAb) immunotherapy in companion animals, one must account for the fact that the Fc region of the MAb, the part not involved in antigen recognition, is typically derived from mice or human protein sequences and thus is considered foreign by the immune system. Hence, repeated treatment with a non–canine-based antibody leads to a robust inactivation of the MAb, as well as systemic toxicity, thus eliminating its effectiveness over time. Forty years ago, Thomas and Burnet, while studying how lymphocytes could respond to newly formed antigens on transformed cells, put forth the concept that the immune system could actively respond to and eliminate cancerous cells, an idea known as immune surveillance.2 In contrast, later studies3,4 showed that genetically manipulated immunodeficient (athymic) mice did not demonstrate an increased incidence of cancer—either spontaneously or carcinogen induced. Such observations were proposed to be due to residual immunity, leading to the immune surveillance concept falling out of favor. Since the development of more sensitive and sophisticated technologies, many of the ideas behind the concept of the immune surveillance hypothesis are now more accepted, and currently, this modification of the original hypothesis is referred to as the immunoediting hypothesis.5 Pivotal studies by Robert Schreiber’s group showed that lymphocytes (T-cells) could directly or indirectly through production of a cytokine, interferon γ (IFN-γ), protect mice against the development of methylcholanthrene (MCA)-induced sarcomas.6,7 Moreover, they demonstrated that tumors from immunodeficient mice were more immunogenic than tumors from immunocompetent mice, thus leading to the immunoediting hypothesis.7,8 This hypothesis consists of three phases: (1) elimination: removal of the immunogenic tumor cells by the immune system; however, weakly immunogenic cells can survive; (2) equilibrium: tumor growth and immune destruction are equal; and (3) escape: tumor growth ensues as the result of decreased immunogenicity, immune suppression, and rapid tumor cell growth.5 However, despite recent data, there is still a controversy that remains around the immune surveillance hypothesis, discussed in a review by Schreiber et al.5 Given the fact that cancer can develop in immunocompetent individuals, some tumor cells are able to avoid recognition by the immune system.9 This is accomplished by various mechanisms (discussed later) that involve both changes in the tumor cells themselves and ways in which the tumor and the tumor stromal environment can manipulate the immune system and prevent antitumor immunity. These mechanisms of immune evasion pose a significant challenge to the development of effective immunotherapies. Figure 13-1 demonstrates some of these key mechanisms. One population of immune cells that play a major role in tumor immunosuppression are myeloid-derived suppressor cells (MDSCs). These cells consist of immature monocytes and granulocytes released from the bone marrow into the blood during pathologic conditions, including cancer. Sometimes included in the functional description of this group of cells are tumor-associated macrophages (TAMs), which have the same ability and use similar mechanisms as MDSCs to induce potent tumor immunosuppression.10,11 Numerous studies demonstrate increased numbers of MDSCs in humans with cancer12–14 and in mouse models of cancer.15,16 Furthermore, it has been shown that the presence of these cells correlates with clinical disease stage and metastatic tumor burden in humans with solid tumors.13 MDSCs are recruited to the tumor microenvironment through various chemoattractants,10 many of which are induced by the tumor during times of hypoxia and are directly released by the effects of hypoxia-inducible factor-1α (HIF-1α) production.17–20 Once at the tumor, MDSCs intercalate into the microenvironment and actively suppress the local antitumor immune response and promote tumor invasion and metastasis via the production of matrix metalloproteinases (MMPs) and various chemoattractants.21,22 Moreover, these cells, including the TAMs, contribute to increased tumor vessel growth, a process referred to as angiogenesis.10 Of note, although cell surface markers are used to identify these cells in mice and humans, similar markers have yet to be discovered for dogs, thus the role that MDSCs play in canine cancer is currently unknown. The ability of MDSCs to suppress the antitumor response is the subject of many recent studies.23–25 Numerous mechanisms of suppression have been reported, and MDSCs have the ability to suppress not only T-cells, but also natural killer (NK) cells and dendritic cells (DCs) and they also are able to potentiate T-regulatory cells (Tregs), which are discussed later, and TAMs in the tumor (see Figure 13-1). Currently known mechanisms of immune suppression by MDSCs include suppression of T-cells through production of inducible nitric oxide (NO) species (iNOS), reactive oxygen species (ROS) and arginase, as well as cysteine deprivation.23 MDSCs can produce transforming growth factor-β (TGF-β) and IL-10, which stimulate Tregs and TAMs, and MDSCs can cause downregulation of the IL-12 production by TAMs, a cytokine involved in T-cell activation.24 MDSCs cause NK cell anergy (lack of function) also by this decreased IL-12 production and through membrane-bound TGF-β. 24,25 Thus, given the ability of these cells to use multiple pathways to induce tumor immunosuppression, the development of effective immunotherapies that can target these cells and either eliminate them or lead to their maturation, rather than ones that target specific pathways of suppression, is critical for that therapy’s success. Another population of cells that are significantly increased in tumor-bearing humans and animals are Tregs. These cells are phenotypically defined by surface expression of CD4 and CD25 but are most specifically identified by the intracellular transcription factor, forkhead box P3 (foxp3).26,27 Other surface markers used to characterize these cells have been described, including cytotoxic T-lymphocyte antigen-4 (CTLA-4), glucocorticoid-induced tumor necrosis factor (TNF) receptor family-regulated gene (GITR), lymphocyte-activation gene 3 (Lag3), and folate receptor-4 (FR-4).28–31 This distinct subset of CD4+ T-cells is capable of directly suppressing tumor-specific CD4+ and CD8+ T-cells and NK cells and are enriched in the tumor microenvironment by conversion of CD4 T-cells to Tregs by DCs or TGF-β,32–35 proliferation of tumor-specific Tregs following antigen recognition, or recruitment of these cells via chemokine signaling (i.e., CCR5).36 Recent work has also suggested a role for the chemokine CCL-1 in specifically converting T-cells to Tregs and inducing their suppressive nature.37 Many studies demonstrate that increased numbers of Treg cells are correlated with a poor prognosis.38–41 Additionally, Tregs present in metastatic lymph nodes inhibit the ability of tumor-infiltrating lymphocytes to mount an effective antitumor response.42 Work in our laboratory demonstrated that canine Treg cells can also be identified via the expression of CD4 and foxp3.43 Moreover, we saw that cancer-bearing dogs had increased numbers of Tregs compared to healthy dogs and that this difference was greater in certain types of canine cancers.43,44 Therefore current therapies aimed at depleting Treg cells in humans could be applied to veterinary medicine. In particular, many studies have shown that the use of cyclophosphamide or anti–Treg-specific antibodies decreases the numbers of Tregs present in tumors and in circulation of tumor-bearing patients.45–49 Another important mechanism of tumor suppression is through impairment of the potent antigen-presenting cells, dendritic cells (DCs). Numerous studies have denoted that overall numbers of DCs are decreased in various human cancers studied, including head and neck squamous cell carcinomas (HNSCCs),50 breast and prostate cancers, and malignant gliomas.51 A recent study showed that indoleamine 2,3-dioxygenase 1 (IDO1) expression in the tumor microenvironment led to increased DC apoptosis.52 Some tumor studies also demonstrated fewer circulating myeloid DCs and a concurrent increase in immature DCs (iDCs) that reduce presentation of antigens and stimulation of T-cells; thus they induce T-cell tolerance rather than activate T-cells.51,53,54 Thus the DCs present in the tumor tend to be immature and dysfunctional. Studies of DCs in numerous human cancers demonstrate minimal activation, decreased ability to stimulate in an alloreactive fashion, and decreased expression of co-stimulatory molecules.50,51,55–60 A similar study done in dogs with canine transmissible venereal tumors (CTVT) showed that the tumor environment caused downregulation of DC surface markers of activation and major histocompatibility (MHC), as well as decreased endocytic capabilities and decreased allogenic mixed lymphocyte reaction (MLR) responses.61 Possible mechanisms causing the DC dysfunction include the overexpression of the protein S100A9,62 accumulation of triglycerides in the DCs that leads to decreased capacity to present antigen,63 and downregulation of toll-like receptor 9 (TLR9).64 Moreover, factors such as IL-10 and vascular endothelial growth factor (VEGF) can negatively affect DC function and maturation.65,66 Finally, some DCs in the tumor are considered to be regulatory based on low expression of surface markers MHC II, CD86, and CD11c with high expression of co-stimulatory molecules CD80, CD40, CD106, and CD11b. These cells secrete regulatory factors such as IL-10 and NO and inhibit proliferation of naïve CD4+ T cells to antigen presented by mature, functional DCs.67 Overall, the microenvironment of the tumor leads to attraction of immature and regulatory DCs that due to their decreased activation and function can potently inhibit the development of antitumor T-cell responses even when copious amounts of antigen are present. In addition to the suppressive milieu established by tumor-infiltrating cells, the tumor cells themselves are capable of producing immunosuppressive cytokines.68 A few key cytokines produced by tumor cells are IL-10, TGF-β, and tumor necrosis factor-α (TNF-α).68,69 These cytokines act to suppress antitumor T-cell responses and inhibit DC function. IL-10 promotes Treg production and function70 and, in an autocrine and/or paracrine fashion, may potentially affect tumor cell proliferation and survival.71 In human cancer patients, increased levels of serum IL-10 is observed in patients with pancreatic carcinoma and non-Hodgkin’s lymphoma (NHL).72,73 In addition, elevated levels of IL-10 in diffuse large B-cell lymphoma in humans correlate with a poor prognosis.74 TGF-β acts similarly to IL-10 in that it is a potent immunosuppressive cytokine that can potentiate Treg proliferation and function.33,75–77 It can also enhance tumor progression; carcinomas can produce excess TGF-β, which in turn increases epithelial-to-mesenchymal transition, tumor invasion, and metastasis, and inhibit tumor-specific CD8+ T-cells.77 Moreover, tumor-produced TNF-α leads to promotion of tumor cell survival via induction of antiapoptotic proteins.78 Finally, TNF-α has been shown to promote tumor angiogenesis and metastasis and hamper cytotoxic T-cell and macrophage responses.79 One study in veterinary medicine examined a lymph node of a dog with metastatic melanoma. This study revealed an overexpression of IL-10 and TGF-β concurrent with a lack of expression of IL-2, IL-4, or IFN-γ—cytokines typically associated with antitumor immunity—thus demonstrating that tumor immunosuppression occurs in veterinary patients as well.80 For a review of cytokines relevant to tumor immunotherapy, see Table 13-1. Tumor cells are also capable of avoiding immune elimination by failing to be recognized by the immune system in the first place. For example, some tumor cells can downmodulate MHC surface expression to escape recognition by T-cells. MHC class I expression can be lost on tumor cells due to changes in protein synthesis, structure, or allelic loss.81,82 Moreover, defects in antigen processing and presentation can occur that can also lead to decreased MHC expression.81,82 A decrease in class II expression is also observed in certain human hematopoietic cancers, although it should be noted that most tumors are normally MHC class II negative.83,84 Reduced expression of MHC class II has been recently correlated with poor outcome in dogs with B-cell lymphoma.85 In addition, tumor cells can express co-inhibitory surface molecules, such as CD73 and/or PD-L1. CD73 is an ecto-5′-nucleotidase that catalyzes the breakdown of adenosine monophosphate (AMP) to adenosine. When expressed on tumors, this creates a local microenvironment rich in adenosine, which is immunosuppressive.52 The programmed death-1/programmed death ligand-1 (PD-1/PD-L1) axis also plays an immunosuppressive role in cancer. PD-L1 expression on tumors downmodulates antitumor T-cell function86–88 and NK cell89 activity via interaction through the PD-L1 expressed on these immune cells. Thus tumor cells themselves can actively and directly suppress antitumor T-cell responses through such mechanisms as decreased expression of MHC molecules and increased expression of inhibitory molecules. Depletion of Immunosuppressive Myeloid-Derived Suppressor Cells to Allow for Effective Immunotherapy In light of many recent studies, it has become clear that in order to develop an effective immunotherapy, it must be able to overcome, or be combined with other treatments that can overcome, the immunosuppression present in the tumor microenvironment. As mentioned previously, MDSCs are a key component of such immunosuppression. Box 13-1 lists the various potential ways in which MDSCs can be manipulated in order to enhance the effectiveness of immunotherapy. In the 1900s, William Coley observed that cancer patients who developed bacterial infections survived longer than those that did not (reviewed in Richardson and colleagues110). Building on these observations, Coley developed “Coley’s toxins,” which consisted of killed cultures of Streptococcus pyogenes and Serratia marcescens that he gave to patients with inoperable sarcomas. Although with this “vaccine,” Coley saw cure rates of approximately 15%, his therapy was discontinued because of its significant failure rate and intolerable side effects. However, this seminal work laid the foundation for further studies aimed at nonspecific, pan-immune activation to treat cancer through the use of biologic-response modifiers (BRMs). One of the most well-known and clinically utilized BRMs is bacillus Calmette-Guérin (BCG), a live, attenuated strain of Mycobacterium bovis. Currently, in human medicine, BCG is intravesically instilled into the bladder where it is considered to be effective as a means to treat and prevent relapse of noninvasive transitional cell carcinoma (TCC).111,112 One proposed mechanism for its antitumor effects relates to the recruitment of neutrophils and their ability to promote urothelial cell turnover.113 This recruitment most likely relates to the ability of BCG to elicit T-helper 1 (Th1) inflammatory cytokines.114,115 The use of BCG in veterinary medicine is rather limited. Although its efficacy has been tested on numerous forms of canine cancer,116 its use as an immunotherapy in dogs is limited. BCG can be safely instilled into canine bladders,117 but the rate of true superficial bladder cancers is extremely low in dogs as compared to humans.118 Recent uses of BCG in canines include treatment of CTVT in conjunction with vincristine119 or, in combination with human chorionic gonadotropin (hCG; LDI-100), treatment of mast cell tumors (MCTs).120 In this study, response rates for grade I and II MCTs were comparable to single-agent vinblastine but without the myelosuppression. Another BRM that has been studied in human and veterinary medicine is Corynebacterium parvum. In human and dog melanoma studies, C. parvum displayed antitumor activity as an adjunct to surgery.121,122 However, efficacy of C. parvum as an immunotherapy in other canine cancers has been disappointing.123 As a tumor grows, the core may become necrotic as the initial tumor cells are deprived of nutrients. Layered on this necrotic core are tumor cells that exist in an area of hypoxia, which puts them out of reach of blood vessels that can supply them with oxygen. These cells are able to remain viable and pose a challenge to most immunotherapies, chemotherapies, and even small molecule drugs due to their restricted location. Recently, researchers have begun to genetically modify facultative anaerobic bacteria that can penetrate and survive in these regions. In fact, it has been shown that several strains of Salmonella, including S. typhimurium and S. choleraesuis, target tumors following systemic administration. These bacteria penetrate the necrotic core and feed on the dead cells while also emitting natural toxins that will destroy surrounding, viable cells. Using a mouse melanoma model, treatment with VNP20009, an attenuated S. typhimurium, was able to slow tumor growth and specifically target primary tumor and metastatic lesions.124 Although this study showed that the effects were independent of B- and T-cells, possible indirect effects of the Salmonella include production of inflammatory cytokines, such as TNF-α.125 Recently, another proposed mechanism involves the ability of Salmonella to induce melanoma cells to express gap junctions that can interact with DCs and cause bits of tumor cell proteins to be loaded and expressed on the surface of these DCs for presentation to T cells.126 Unfortunately, in human trials, the bacteria failed to colonize some patients and did not provide any antitumor activity.127 Administration of VNP20009 in dogs resulted in a more positive outcome than in humans. In a phase I clinical trial, VNP20009 was administered to dogs with a variety of malignant tumors.128 In this study, 41 dogs received intravenous infusions of VNP20009 either weekly or biweekly at escalating doses. Fever and vomiting were reported as dose-limiting toxicities. Bacterial colonization was seen in approximately 40% of dogs, and significant clinical responses were observed in 15% of patients, with an overall rate of 37% of dogs experiencing either a transient response or stable disease. Thus the use of VNP20009 in specific dog tumors should be further investigated, perhaps in combination with modified Salmonella engineered to deliver tumor cytotoxic agents. Bacteria, such as Staphylococcus aureus, produce enterotoxins known as superantigens (SAgs). Two of these S. aureus enterotoxins, referred to as SEA or SEB, stimulate T-cell proliferation and Th1 cytokine production (IL-2, TNF-α, and IFN-γ) via their ability to cross-link the T-cell receptor and MHC class II molecules. These activated T-cells are highly cytolytic and antitumorigenic in mouse models of cancer.129,130 Moreover, the potency of SAgs is increased when delivered with stimulatory cytokines.131,132 However, when these SAgs were injected into humans, toxic shock syndrome was elicited.133 Therefore genetically modified versions of the SAgs that maintain their immune potency have been evaluated in humans. In a study of patients with non–small-cell lung carcinoma using a modified SAg, stable disease occurred in 42% of patients with decreased tumor burdens of up to 50%. The dose-limiting side effect noted in this study was hypotension.134 SAgs and modified versions are still currently being evaluated for potential use as a human cancer therapeutic.135,136 In veterinary medicine, SAgs were evaluated for efficacy and safety in dogs with oral melanoma and soft tissue sarcoma (STS). Dow et al assessed the efficacy of intratumoral injection of lipid-complexed plasmid DNA that encoded for SEB and either granulocyte-macrophage colony-stimulating factor (GM-CSF) or IL-2.131 Complete or partial remission was seen in 46% of dogs and increased survival time for stage III diseased dogs was observed. There were no toxicities noted, and analysis of tissue sections revealed increased infiltration of tumors with T-cells and macrophages. Thamm et al assessed the efficacy of a lipid-DNA-SEA/IL-2 therapy in dogs with STS.137 In this study, dogs received once weekly intratumoral injections and surgery was done after the 12-week treatment. In the 25% of dogs that responded to the therapy (3 complete responders, 1 partial), a diffuse lymphoplasmacytic infiltrate was observed on histologic evaluation of the tumors. Thus SAgs show some promise for use in veterinary medicine. Similar to SAgs, bacterial cell components such as peptides derived from mycobacterial cell walls were evaluated for potential immunogenicity. One such product is muramyl tripeptide (MTP), which, when encapsulated in a phosphatidylethanolamine-based liposome (L-MTP-PE), can efficiently activate monocytes and macrophages to produce proinflammatory cytokines, such as IL-1α and β, IL-6, IL-7, IL-8, IL-12, and TNF-α.138 The use of L-MTP-PE as a therapeutic was assessed in phase I and II trials of people with osteosarcoma (OSA), renal carcinoma, and metastatic melanoma.138–140 Moreover, this drug has been approved for use in treating pediatric osteosarcoma in Europe under the name Mifamurtide.141 L-MTP-PE has been evaluated in veterinary medicine in a variety of studies.142–146 The survival benefit of L-MTP-PE therapy has been most clearly demonstrated in dogs with appendicular OSA.147 In this study, dogs receiving L-MTP-PE following limb amputation had a median survival time (MST) of 222 days, whereas dogs that received placebo had an MST of 77 days. However, since most of the dogs in both groups developed metastatic disease, further studies evaluated the efficacy of L-MTP-PE in conjunction with chemotherapy.142 In one study, dogs receiving L-MTP-PE after treatment with cisplatin had an MST of 14.4 months versus 9.8 months in dogs that received cisplatin only. Interestingly, only 73% of dogs receiving L-MTP-PE developed metastatic disease compared to 93% in the cisplatin only group. However, in a second trial, these investigators saw no significant survival advantage in dogs with OSA that received L-MTP-PE concurrently with cisplatin. The authors postulated that cisplatin obviated antimetastatic potential of L-MTP-PE due to impaired immune effectors. L-MTP-PE was also evaluated for efficacy in canine hemangiosarcoma (HSA).144 Dogs that received L-MTP-PE with chemotherapy following splenectomy had an MST of 9 months versus the 5.7 months seen with dogs receiving chemotherapy alone. In another study, only dogs with stage I oral melanoma that received L-MTP-PE had an increased survival over placebo-treated dogs.145 No differences were seen within the dogs with more advanced disease. Bacterial DNA can also stimulate the innate immune system via its CpG-oligonucleotides (CpG-ODNs), particularly when complexed with cationic liposomes, in a form known as cationic lipid-DNA complexes (CLDC).148 Complexing bacterial plasmid DNA to liposomes allows for more efficient delivery of the CpG DNA to the endosomal compartment of antigen-presenting cells such as DCs, in which it is released from the liposomes and binds to its receptor, TLR9.149,150 In mouse studies, CLDC stimulates the immune system largely through induction of NK cell activity and release of IFN-γ.148 Moreover, CLDC was also shown to stimulate the production of type I IFN151 and thus is a potent nonspecific immunostimulant. The use of CLDC in dogs has been evaluated in metastatic OSA and in dogs with STS.152,153 Intravenous administration of a modified CLDC that encodes for IL-2 was performed in dogs with stage IV OSA.152 Dogs that received CLDC developed fevers and showed changes in their leukogram profile indicative of immune stimulation. Moreover, NK cell activity was observed, as assessed by target cell lysis, and monocytes showed increased expression of B7.2 on their surface, indicating activation. Treatment was associated with a significant increase in survival times compared to historic controls. Another study examined the use of CLDC in canine STS. Administration of CLDC intravenously once weekly for 6 weeks resulted in an objective response in 15% of the dogs and a decrease in tumor mean vessel density in half of the dogs receiving treatment.153 Thus CLDC has potential to be used as a stand-alone immunotherapeutic in veterinary medicine for a variety of cancer types. Oncolytic viruses are defined as viruses capable of replicating in and lysing tumor cells, thus making them a likely candidate for drug or gene delivery to tumors. A beneficial side effect of these viruses is that they can kill the tumor cell, thus providing release of TAs for processing by the immune system. Adenoviruses that have undergone genetic modification of their early genes, 1A (E1A) and 1B (E1B), preferentially target rapidly dividing tumor cells and have been used to target canine OSA cells.154–156 Canine distemper virus (CDV) has also been investigated as a treatment for B- and T-cell lymphoma in dogs.157 In vitro studies using fluorescently labeled, attenuated CDV and canine lymphoma cells demonstrated that CDV infected lymphoid cells via binding of the cell membrane protein CD150, which is overexpressed on malignant B cells, and induced cellular apoptosis.157 The therapeutic use of IL-2 in humans is fraught with toxicity.158–160 However, the use of IL-2 therapy in veterinary medicine holds some promise as a therapeutic. First, Helfand et al demonstrated that intravenously injected recombinant human IL-2 (rhIL-2) activates canine lymphocytes, causing only mild gastrointestinal toxicity, even at high doses for 4 consecutive days.161 Another study demonstrated the ability of rhIL-2 to induce canine lymphokine-activated killer (LAK) cells and incidentally showed that LAK cells from tumor-bearing dogs did not kill tumor cells as efficiently as compared to normal dogs.162 Further evaluation of toxicity and efficacy of rhIL-2 was done using dogs with primary lung cancer and with lung metastases in an aerosol formulation.163 In this study, complete regression was seen in two of the four dogs with pulmonary metastases, and these dogs remained disease free for at least 12 months after treatment. One of the two dogs with a primary lung tumor had disease stabilization for more than 8 months, whereas the other dog had progressive disease. Assessment of the lymphocytes obtained from bronchoalveolar lavage showed increased cytolytic activity following 15 days of IL-2 treatment. In addition, minimal toxicity was noted in this study. Finally, IL-2 gene therapy using viral vectors has been examined for treatment of feline fibrosarcomas and canine melanoma and was shown to be safe and effective.164–166 Therefore, given its low toxicity and promising effectiveness, rhIL-2 therapy is a plausible treatment for canine cancer. IL-12, produced by antigen-stimulated DCs, macrophages, and B-cells, plays a role in stimulating the growth and function of T-cells and enhances the cytolytic activity of both T-cells and NK cells. Similar to IL-2, IL-12 therapy in humans leads to serious side effects; currently, it is not used clinically. Current investigation into the use of IL-12 in veterinary medicine revolves around recombinant gene therapy for treatment of canine head and neck tumors,167 with some in vitro work looking at its use in feline hyperthermia-induced gene therapy.168 IL-15 is structurally similar to and uses similar signaling molecules as IL-2. IL-15 plays a role in stimulation of NK cells and promoting proliferation of T-cells. However, from an immunotherapy standpoint, IL-15 holds more promise than IL-2 in that (1) it does not cause activation-induced cell death of CD4+ T-cells following prolonged periods of exposure; rather it sustains T cell proliferation,169 (2) IL-15 plays a critical role in CD8+ T-cell memory formation and maintenance,170 and (3) unlike IL-2, IL-15 does not appear to play a role in the development of Tregs.1 Clinical investigation of IL-15 will begin soon. An initial safety study in nonhuman primates was recently conducted.171 Twelve daily doses of clinical grade rhIL-15 revealed that neutropenia was the dose-limiting toxicity and documented an increase in circulating NK cells and memory CD8+ T-cells. In veterinary medicine, one study used plasmid IL-15 in combination with plasmid IL-6 in beagles with CTVT.172 A threefold increase in the proportion of CD8+ T-cells that infiltrated the tumors and an enhancement of IFN-γ–producing cells and increased cytolytic activity against the tumor were observed. Thus IL-15 therapy shows promise as an effective immunotherapy in both human and veterinary medicine. Type I IFNs can affect cellular proliferation through various mechanisms, including interactions with cell cycle proteins (i.e., c-myc and retinoblastoma) and induction of apoptosis via Bcl-2/Bax and TNF/Fas interactions. Their antiangiogenic properties of downregulating VEGF and basic fibroblast growth factor (bFGF)173 make them attractive as immunotherapies, and they have been used successfully to treat pediatric hemangiomas.174 Clinical trials using type I IFNs have met limited success due to the high occurrence of severe toxicity in light of overall limited response rates. Nonetheless, their effectiveness was assessed in melanoma, multiple myeloma, renal cell carcinoma, leukemia, and other cancers, as well as in conjunction with chemotherapies. The best response, in terms of disease-free survival, was seen in renal cell carcinoma and melanoma when used as single agents.175,176 The use of type I IFNs in veterinary medicine is limited and mostly used for feline viral therapies.177 One study showed that recombinant feline IFN-ω was safe and easy to use for treating feline fibrosarcomas. As this was a safety study, the therapeutic effects of this treatment were not evaluated. Another recently published study also used recombinant feline IFN-ω with or without chemotherapy to study its effects in treating mammary tumors in vitro.178 This study reported that the antitumor cell effects of recombinant IFN and chemotherapy were additive and suggested further investigation into its clinical use as an adjuvant therapy.
Cancer Immunotherapy
Immune System Control of Tumor Development and Growth
Mechanisms of Immune Evasion by Tumors
Active Immune Suppression by Myeloid-Derived Suppressor Cells
Induction of Regulatory T-Cells by Tumors
Impaired Dendritic Cell Activation and Function
Production of Immunosuppressive Cytokines
Failure of Tumor Cells to Activate Immune System
Strategies to Control Tumor Growth through Immune Activation
Nonspecific Immune Activation to Generate Antitumor Activity Using Biologic-Response Modifiers
Bacillus Calmette-Guérin and Corynebacterium parvum
Salmonella
Superantigens
Liposome-Encapsulated Muramyl Tripeptide
Liposome-DNA Complexes
Oncolytic Viruses
Nonspecific Tumor Immunotherapy Using Recombinant Cytokine Therapy
Interleukin-12
Interleukin-15
Interferons
Interferon-α, Interferon-β, and Interferon-ω
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cancer Immunotherapy