Chapter 14 BETA-CELL NEOPLASIA: INSULINOMA
Insulin-secreting beta-cell tumors were first described in the dog by Slye and Wells in 1935. During the past 68 years, numerous publications have appeared in the veterinary literature addressing the clinical manifestations, diagnosis, treatment, and pathology of beta-cell tumors in dogs. Despite excellent documentation of this disease and well-established methods for determining the diagnosis, insulin-secreting beta-cell neoplasia remains an uncommon diagnosis in dogs and a rare entity in cats.
ETIOLOGY
Functional tumors arising from the beta cells of the pancreatic islets are malignant tumors that secrete insulin independent of the typically suppressive effects of hypoglycemia. Beta-cell tumors, however, are not completely autonomous, and they respond to provocative stimuli (e.g., glucose) by secreting insulin, often in excessive amounts. Immunohistochemical analysis of beta-cell tumors has revealed a high incidence of multihormonal production, including pancreatic polypeptide, somatostatin, glucagon, serotonin, and gastrin (Hawkins et al, 1987; O’Brien et al, 1987; Minkus et al, 1997). However, insulin has been the most common product demonstrated in the neoplastic cells, and the clinical signs in such animals are primarily those that result from a hyperinsulinemia-induced hypoglycemia.
MALIGNANT VERSUS BENIGN POTENTIAL.
Beta-cell tumors are notorious for masking their malignant tendencies in the dog. Discrepancy is noted between the orderly arrangement of well-differentiated cells, the rarity of the mitotic figures seen in most islet cell tumors, and the frequent metastasis of beta-cell tumors (Kruth et al, 1982). Differentiation of malignant from benign neoplasia is usually based on identification of metastasis at surgery or necropsy, or the recurrence of hyperinsulinism weeks to months after surgical removal of a “solitary” pancreatic mass. Image analysis techniques applied to tumor tissue have also shown promise for differentiating between metastatic and nonmetastatic islet cell tumors (Minkus et al, 1997).
PATHOPHYSIOLOGY
Maintenance of euglycemia in the healthy dog
Cell metabolism and function depend on the delivery of energy sources via the circulation. Exogenously derived energy, in the form of ingested carbohydrate, fat, and protein, provides enough fuel for 4 to 8 hours of cell metabolism. After this postprandial period, fuel for cellular metabolism must be derived from endogenous sources, primarily through production of glucose by the liver (Fig. 14-1). The liver initially provides glucose by the breakdown of stored hepatic glycogen (glycogenolysis). Liver glycogen stores are exhausted slowly in dogs, requiring 2 to 3 days of fasting, compared with only 24 hours of fasting in humans (de Bruijne, 1982). Hepatic glucose production is augmented by gluconeogenesis as the postprandial period increases and hepatic glycogen stores become depleted (Rothman et al, 1991). Gluconeogenesis is the formation of glucose from precursors, including amino acids (especially alanine and glutamine), lactate, and glycerol. These substrates are delivered to the liver from peripheral stores. Muscle and other structural tissue supply amino acids, mainly alanine; blood cell elements supply lactate, the end product of glycolytic metabolism; and adipose tissue supplies glycerol from lipolysis of triglycerides (Karam, 2001). Oxidation of free fatty acids released from adipose cells during lipolysis supplies the energy required for gluconeogenesis and provides ketone bodies (i.e., acetoacetate, β-hydroxybutyrate), which can serve as alternative metabolic fuels for the central nervous system (CNS) during periods of prolonged fasting. Fatty acids are believed to be the major mediator of hepatic gluconeogenesis (Karam, 2001).
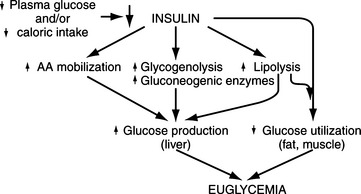
Hepatic glucose production depends on an adequate supply of substrates, including certain amino acids, glycerol, and free fatty acids mobilized from muscle and adipose tissue (Cryer and Polonsky, 1998). Other requirements include a normal hepatic circulation, functioning hepatocytes capable of removing substrates from the circulation, and a complete complement of hepatic enzymes capable of converting these noncarbohydrate precursors into glucose.
The renal cortex also has the requisite enzymes for the production and release of glucose into the circulation, albeit the contribution is only about 5% during fasting (Stumvoll et al, 1995; Gerich et al, 2001). However, renal glucose production is regulated, and under certain circumstances (e.g., glucose counterregulation, hepatic insufficiency), the contribution of glucose derived from renal gluconeogenesis can be as high as 40%. The kidney does not have glycogen stores and depends on gluconeogenesis as its only source of glucose production. Glutamine rather than alanine is the predominant amino acid substrate for renal gluconeogenesis. In addition to its contribution to glucose homeostasis during fasting, the kidney has been shown to be an important contributor to glucose counterregulation in the event of hypoglycemia. Although glucagon does not affect the kidney, the counterregulatory increase in epinephrine has been shown to stimulate gluconeogenesis in the renal cortex (Stumvoll et al, 1995; Gerich et al, 2001).
A normally functioning endocrine system is also necessary to maintain glucose homeostasis and prevent hypoglycemia. Insulin is the dominant glucose-lowering hormone. It suppresses endogenous glucose production and stimulates glucose utilization. Glucose-raising or counterregulatory hormones include glucagon, epinephrine, growth hormone, and cortisol. These hormones increase hepatic glucose production by stimulating glycogenolysis and gluconeogenesis. They also inhibit glucose utilization by tissues (Cryer and Polonsky, 1998).
Hypoglycemia and the counterregulatory response
Identification of a randomly obtained or fasting blood glucose concentration below 60 mg/dl is cause for concern in either dogs or cats. Although this finding is not always diagnostic of organic disease, “normal” dogs and cats have consistently been shown to not have blood glucose concentrations decline below this level. Therefore, once artifactual error has been eliminated as a possible cause, organic disease is a likely cause of persistent hypoglycemia (see page 623). Failure in any one of the steps involved in hepatic glucose production may result in hypoglycemia and its related clinical signs. Abnormalities affecting any of the key steps involved in the production and conservation of glucose may also cause hypoglycemia.
Under the usual metabolic conditions, the CNS is wholly dependent on plasma glucose and counteracts declining blood glucose concentrations with a carefully programmed neurogenic and hormonal response to mobilize storage depots of glycogen and fat and raise the plasma glucose concentration (Tables 14-1 and 14-2). Hepatic glycogen reserves and gluconeogenesis in the liver and kidney directly supply the CNS with glucose, and the mobilization of fatty acids from triglyceride depots provides energy for the large mass of skeletal and cardiac muscle, the renal cortex, the liver, and other tissues that use fatty acids as basic fuel; this spares glucose for use by tissues that remain dependent on glucose, including the CNS, erythrocytes, bone marrow, and renal medulla (Karam, 2001). The cascade of events leading to endogenous glucose production is initiated by a decreasing blood glucose concentration that eventually declines below a critical level (approximately 65 mg/dl). When this occurs, the autonomic nervous system and counterregulatory hormone response are stimulated to restore an adequate supply of glucose for cerebral function.
TABLE 14-1 AUTONOMIC NERVOUS SYSTEM RESPONSE TO HYPOGLYCEMIA
Adapted from Karam JH: Hypoglycemic disorders. In Greenspan FS, Gardner DG (eds): Basic and Clinical Endocrinology, 6th ed. New York, Lange Medical Books/McGraw-Hill, 2001, p 702. (source)
TABLE 14-2 INSULIN AND COUNTERREGULATORY HORMONAL RESPONSE TO HYPOGLYCEMIA
Hormones are the most important glucoregulatory factors, and glucose is the most important determinant of the secretion of glucoregulatory hormones. Insulin is the dominant glucose-lowering hormone. Hypoglycemia suppresses insulin secretion, which facilitates the mobilization of energy from existing energy stores (glycogenolysis, lipolysis), promotes hepatic gluconeogenesis and ketogenesis, promotes renal gluconeogenesis, and decreases glucose utilization by insulin-dependent tissues (Cryer and Polonsky, 1998; Karam, 2001). Glucose-raising or counterregulatory hormones include glucagon, epinephrine, growth hormone, and cortisol (see Table 14-2). Insulin-induced hypoglycemia causes an increase in plasma glucagon, epinephrine, and norepinephrine concentrations at the onset of the glucose counterregulatory response, with increases in plasma growth hormone and cortisol occurring later. Glucagon is the key counterregulatory hormone affecting recovery from acute hypoglycemia, although its role in recovery from chronic hypoglycemia appears to be less influential (Cryer and Gerich, 1985). In response to falling plasma glucose levels, glucagon is secreted by the alpha cells of the pancreatic islets into the hepatic portal circulation; it acts exclusively on the liver to activate glycogenolysis and gluconeogenesis (Cryer and Polonsky, 1998). Hepatic glucose production increases within minutes.
The adrenergic-catecholamine response to hypoglycemia also plays a major role in recovery from hypoglycemia. Epinephrine has both direct and indirect effects, which stimulate hepatic glycogenolysis and hepatic and renal gluconeogenesis; provide muscle tissue with an alternative source of fuel by mobilizing muscle glycogen and stimulating lipolysis; mobilize gluconeogenic precursors (e.g., lactate, alanine, and glycerol); and inhibit glucose utilization by insulin-sensitive tissues (e.g., skeletal muscle) (Cryer, 1993; Karam, 2001). The role of cortisol and growth hormone in the acute response to hypoglycemia is probably minimal. Longer term elevation of cortisol facilitates lipolysis, promotes protein catabolism and the conversion of amino acids to glucose by the liver and kidney, and limits glucose utilization by insulin-dependent tissues. Similarly, growth hormone promotes lipolysis and antagonizes the action of insulin on glucose utilization in muscle cells. However, the hyperglycemic effects of cortisol and growth hormone do not appear for several hours after the hypoglycemic episode (Cryer and Polonsky, 1998). Cortisol and growth hormone play roles in the defense against prolonged hypoglycemia (Boyle and Cryer, 1991).
The adrenergic neurogenic response to hypoglycemia acts directly to raise the blood glucose concentration and to stimulate hormonal responses that augment the adrenergic mobilization of energy stores (see Table 14-1). In dogs, hepatic glucose autoregulation is also an important glucose counterregulatory factor (Cryer and Polonsky, 1998). That is, the rate of hepatic glucose production is an inverse function of the blood glucose concentration independent of hormonal and neural regulatory factors.
Regulation of insulin secretion in the healthy dog
The ratio between the blood insulin and glucose concentration appears to remain constant, even during a prolonged fast (de Bruijne et al, 1981). The beta cells of the pancreatic islets are primarily responsible for monitoring and controlling the blood glucose concentration. Unlike with most other cells, the entrance of glucose into beta cells occurs independent of insulin. When blood glucose concentrations exceed approximately 110 mg/dl, the insulin secretion rate increases and the blood glucose concentration declines. When blood glucose concentrations decrease below 60 mg/dl, insulin secretion is inhibited, which limits continued tissue utilization of glucose and increases the blood glucose concentration.
Insulin secretion in dogs with beta-cell neoplasia
Failure of insulin secretion to decrease during periods of hypoglycemia predisposes a dog with a beta-cell tumor to the development of clinical signs of hypoglycemia during fasting and exercise. Insulin-secreting beta-cell tumors also remain responsive to many of the stimuli that promote insulin secretion in the healthy dog, but the secretory response is often exaggerated, resulting in severe hypoglycemia. For example, clinical signs of hypoglycemia often occur after consumption of food or intravenous administration of glucose to correct hypoglycemia (see page 621).
Mechanism for insulin-induced hypoglycemia
Insulin-secreting tumors and the associated hyperinsulinemia interfere with glucose homeostasis by decreasing the rate of glucose release from the liver and increasing the utilization of glucose by insulin-sensitive tissues (e.g., muscle, adipose tissue). Insulin interferes with mechanisms that promote hepatic glucose output by limiting circulating concentrations of substrates needed for gluconeogenesis. This effect is accomplished by inhibiting enzymes necessary for mobilizing amino acids from muscle and glycerol from adipose tissue. In addition, insulin decreases the activity of hepatic enzymes used in gluconeogenesis and glycogenolysis. Insulin also lowers blood glucose concentrations by stimulating glucose uptake and utilization in the liver, muscle, and adipose tissue. Thus insulin increases tissue utilization of glucose already present in the extracellular space while interfering with hepatic production of glucose. The net effect is decreasing blood glucose concentrations because of increased tissue utilization of glucose (see Feldman and Nelson, 1987 for references).
Origin of clinical signs
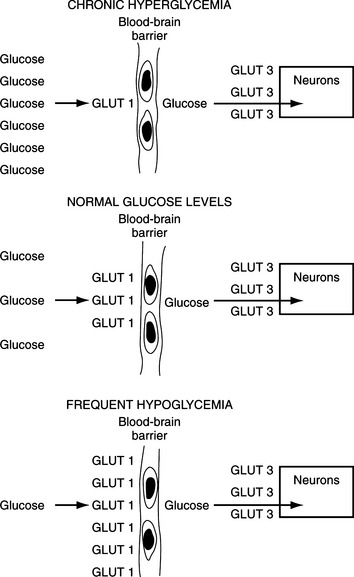
The entrance of glucose into the neurons of the CNS occurs primarily by diffusion and is not insulin dependent. Because cell membranes are impermeable to hydrophilic molecules such as glucose, all cells require carrier proteins to transport glucose across the lipid bilayers into the cytosol. All cells except those in the intestine and kidney have non-energy-dependent transporters that facilitate diffusion of glucose across cell membranes. At least five “facilitative glucose transporters” have been described in humans, and these are called GLUT 1 through GLUT 5, with the numbers designating the order of their identity (Table 14-3) (Masharani and Karam, 2001). GLUT 1 is present in all tissues, has a very high affinity for glucose, and appears to mediate basal glucose uptake. GLUT 1 is an important component of the brain vascular system (blood-brain barrier) that ensures adequate transport of plasma glucose into the CNS (Fig. 14-2). GLUT 3 is the major glucose transporter on the neuronal surface, has a very high affinity for glucose, and is responsible for transferring glucose from the cerebrospinal fluid into the neuronal cells.
TABLE 14-3 GLUCOSE TRANSPORTERS IDENTIFIED IN HUMANS
| Name | Major Sites of Expression | Affinity for Glucose* |
|---|---|---|
| GLUT 1 | Brain vasculature, red blood cells, all tissues | High (Km = 1 mmol/L) |
| GLUT 2 | Liver, pancreatic B cells, serosal surfaces of gut and kidney | Low (Km = 15–20 mmol/L) |
| GLUT 3 | Brain neurons, also found in all tissues | High (Km < 1 mmol/L) |
| GLUT 4 | Muscle, fat cells | Medium (Km = 2.5 to 5 mmol/L) |
| GLUT 5 | Jejunum, liver, spermatozoa | Medium (Km = 6 mmol/L) |
* Km represents the level of blood glucose at which the transporter has reached one-half of its maximum capacity to transport glucose. It is inversely proportional to the affinity.
From Masharani U, Karam JH: Pancreatic hormones and diabetes mellitus. In Greenspan FS, Gardner DG (eds): Basic and Clinical Endocrinology, 6th ed. New York, Lange Medical Books/McGraw-Hill, 2001, p 630.
Blood insulin concentrations do not affect neuronal glucose transport or utilization. However, if hyperinsulinemia results in an inadequate glucose supply for intracellular oxidative processes in neurons, a resultant decline occurs in energy-rich phosphorylated compounds (adenosine triphosphate [ATP]) in neurons. This in turn results in cellular changes typical of hypoxia, increased vascular permeability, vasospasm, vascular dilation, and edema. Neuron death from anoxia follows. In acute hypoglycemia, histologic alterations are most marked in the cerebral cortex, basal ganglia, hippocampus, and vasomotor centers (see Feldman and Nelson, 1987 for references). Although most of the damage from hypoglycemia occurs in the brain, peripheral nerve degeneration and demyelination are sometimes encountered (Braund et al, 1987). Other major organ systems, such as the heart, kidneys, and liver, also depend on glucose. However, an acute decrease in the blood glucose concentration results in clinical signs that involve the CNS before signs of any other major organ system dysfunction become apparent.
Prolonged, severe hypoglycemia may result in irreversible brain damage; however, it is uncommon for a dog to die during a hypoglycemic episode. Hypoglycemia is a potent stimulus for the release of the counterregulatory hormones that function to antagonize the effects of insulin and stimulate an increase in the blood glucose concentration (see Hypoglycemia and the Counterregulatory Response, page 617).
The clinical manifestations of hypoglycemia are believed to result from both a lack of glucose supply to the brain (neuroglycopenia) and stimulation of the sympathoadrenal system. The neuroglycopenic signs common to dogs include lethargy, weakness, ataxia, bizarre behavior, convulsions, and coma (Table 14-4). Clinical signs resulting from stimulation of the sympathoadrenal system include muscle tremors, nervousness, restlessness, and hunger. In humans, the symptoms related to release of catecholamines often precede those of neuroglycopenia and act as an early warning sign of an impending hypoglycemic attack (Karam, 2001). This illustrates the rapid response of catecholamine secretion to hypoglycemia and partly explains why canine patients with insulin-secreting tumors do not always progress to generalized seizure activity during a fast.
TABLE 14-4 CLINICAL SIGNS ASSOCIATED WITH INSULIN-SECRETING TUMORS IN 91 DOGS
| Clinical Sign | Number of Dogs | Percent |
|---|---|---|
| Seizures | 51 | 56 |
| Weakness | 43 | 47 |
| Collapse | 27 | 30 |
| Ataxia | 17 | 19 |
| Muscle fasciculations | 16 | 18 |
| Posterior weakness | 15 | 16 |
| Depression, lethargy | 15 | 16 |
| Bizarre behavior | 11 | 12 |
| Polyphagia | 6 | 7 |
| Polyuria, polydipsia | 6 | 7 |
| Weight gain | 6 | 7 |
| Diarrhea | 4 | 4 |
| Syncope | 3 | 3 |
| Nervousness | 3 | 3 |
| Head tilt | 3 | 3 |
| Anorexia | 3 | 3 |
| Urinary incontinence | 2 | 2 |
| Blindness | 2 | 2 |
| Panting | 1 | 1 |
Clinical manifestations depend on the duration and severity of hypoglycemia. Animals with chronic and/or recurring fasting hypoglycemia appear to tolerate low blood glucose levels (i.e., 20 to 30 mg/dl) for prolonged periods without exhibiting clinical signs. In these dogs, slight reductions in the blood glucose level often produce symptomatic episodes. The “adaptation process” to chronic severe hypoglycemia is believed to involve “up-regulation” of the high-affinity glucose transporter GLUT 1 on the vascular cells forming the blood-brain barrier (see Fig. 14-2) (Karam, 2001).
CLINICAL FEATURES
Signalment
Insulin-secreting tumors typically occur in middle-aged or older dogs. The mean age at the time of diagnosis of an insulin-secreting tumor in 97 dogs in our series was 9.8 years, with a median age of 10 years and an age range of 3 to 14 years (Fig. 14-3). There is no gender predilection. A variety of breeds have been diagnosed with an insulin-secreting tumor at our hospital (Table 14-5). Labrador Retrievers, Golden Retrievers, and German Shepherd dogs are the breeds most commonly diagnosed with this disease, which is probably a reflection of breed popularity in our region rather than a breed predisposition per se. In general, insulin-secreting tumors occur more commonly in large breeds of dogs. However, the size of the dog should never preclude an investigation for an insulin-secreting tumor in a hypoglycemic dog. We have diagnosed insulin-secreting tumors in dogs as small as a Pomeranian.
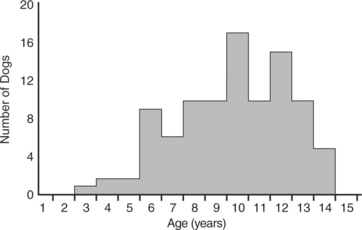
FIGURE 14-3 Age of 97 dogs at the time of initial diagnosis of an insulin-secreting islet cell tumor.
TABLE 14-5 BREED DISTRIBUTION OF 89 DOGS WITH ISLET CELL TUMORS
| Breed | Number of Dogs | Percent |
|---|---|---|
| Labrador Retriever | 12 | 13 |
| Golden Retriever | 10 | 11 |
| German Shepherd | 7 | 8 |
| Irish Setter | 6 | 7 |
| Terriers (Fox, Kerry Blue, West Highland White, Norwich) | 6 | 7 |
| Poodle | 6 | 7 |
| Collie | 5 | 6 |
| Cocker Spaniel | 5 | 6 |
| Mixed-breeds | 5 | 6 |
| Doberman Pinscher | 3 | 3 |
| Boxer | 3 | 3 |
| Border Collie | 3 | 3 |
| Rottweiler | 3 | 3 |
| Samoyed | 2 | 2 |
| Other breeds (1 dog each) | 13 | 15 |
History
The clinical signs of an insulin-secreting tumor typically are caused by neuroglycopenia (hypoglycemia) and an increase in circulating catecholamine concentrations; the signs include seizures, weakness, collapse, ataxia, muscle fasciculations, and bizarre behavior (see Table 14-4). One characteristic of hypoglycemic signs, regardless of the cause, is their episodic nature. Signs are generally observed intermittently for only a few seconds to minutes because of the compensatory counterregulatory mechanisms that usually increase the blood glucose concentration after the development of hypoglycemia. If these mechanisms are inadequate, seizures may occur as the blood glucose concentration continues to decrease. Seizures are usually self-limiting, typically lasting from 30 seconds to 5 minutes. The seizure may stimulate further catecholamine secretion and other counterregulatory mechanisms that increase the blood glucose level above critical concentrations.
Physical Examination
Physical examination findings in dogs with an insulin-secreting tumor are often surprisingly unremarkable, especially if clinical signs of hypoglycemia have been present for several months prior to presentation. Most abnormalities identified on the physical examination are nonspecific and supportive of a diagnosis of hypoglycemia (Table 14-6). Weakness and lethargy, the most common findings, are identified in approximately 40% and 20% of our cases, respectively. Episodes of collapse and seizures may occur during the examination but are uncommon. Afflicted dogs are usually free of palpable abnormalities, aside from findings commonly associated with aging (e.g., lipomas). Weight gain is evident in some dogs and is probably a result of insulin’s potent anabolic effects. Failure to identify abnormalities on the physical examination, especially in an older, large-breed dog, is an important finding supportive of an insulin-secreting tumor.
TABLE 14-6 PHYSICAL EXAMINATION FINDINGS ASSOCIATED WITH INSULIN-SECRETING TUMORS IN 52 DOGS
| Physical Examination Finding | Number of Dogs | Percent |
|---|---|---|
| Weakness | 22 | 42 |
| Lethargy | 10 | 19 |
| Collapsing | 6 | 12 |
| Seizures | 4 | 8 |
| Tremors | 4 | 8 |
| Obtunded | 4 | 8 |
| Peripheral neuropathy | 3 | 6 |
| Obese | 2 | 4 |
| No abnormalities identified | 24 | 46 |
PERIPHERAL NEUROPATHY.
Peripheral neuropathies have been reported in dogs with insulin-secreting tumors (Shahar et al, 1985; Braund et al, 1987; van Ham et al, 1997). Clinical signs and physical examination findings range from paraparesis to tetraparesis, facial paresis to paralysis, hyporeflexia to areflexia, hypotonia, and muscle atrophy of the appendicular, masticatory and/or facial muscles. Sensory nerves may also be affected. A subclinical polyneuropathy has also been reported (Braund et al, 1987). The onset of clinical signs may be acute (days) or insidious (weeks to months). Abnormalities identified on electrodiagnostic testing include abnormal spontaneous potentials (e.g., positive sharp waves, fibrillation potentials) and slowed motor nerve conduction velocities (Braund et al, 1987). Cerebrospinal fluid (CSF) analysis produces a normal result (van Ham et al, 1997). Histopathologic findings in motor and sensory nerves include moderate to severe axonal necrosis, nerve fiber loss, and variable demyelination-remyelination (Braund et al, 1987; Schrauwen et al, 1996; van Ham et al, 1997). Muscle changes reflect neurogenic atrophy. The pathogenesis of the polyneuropathy is not known. Proposed theories include metabolic derangements of the nerves induced by chronic and severe hypoglycemia or some other tumor-induced metabolic deficiency, an immune-mediated paraneoplastic syndrome resulting from shared antigens between tumor and nerves, or toxic factors produced by the tumor that deleteriously affect the nerves (Kudo and Noguchi, 1985; Das and Hochberg, 1999; Heckmann et al, 2000). Treatment is aimed at surgical removal of the beta-cell tumor (Jeffery et al, 1994). Prednisone therapy (initially 1 mg/kg daily) may also improve clinical signs (van Ham et al, 1997). Correction of hypoglycemia, by itself, may not improve clinical signs caused by the peripheral neuropathy (Bergman et al, 1994).
CLINICAL PATHOLOGIC ABNORMALITIES
Virtually all dogs with insulin-secreting tumors remain undiagnosed and often unsuspected of having such tumors after completion of the history and physical examination. Most afflicted dogs have in common the history of episodic weakness or seizures. These signs encompass a wide variety of organic disorders (Table 14-7). The minimum diagnostic evaluation for dogs with these signs should include a complete blood count (CBC), serum biochemical panel, and urinalysis in an effort to identify abnormalities supportive of one of the disorders outlined in Table 14-7. Therapy other than that required for an emergency situation should be withheld until a diagnosis has been made.
TABLE 14-7 A PARTIAL LISTING OF THE NUMEROUS DISORDERS THAT MAY RESULT IN EPISODIC WEAKNESS (INCLUDES SEIZURES)
Acquired: myasthenia gravis, tetanus, discospondylitis, idiopathic polyradiculoneuritis (Coon Hound paralysis), polymyositis, polyarthritis |
The results of the CBC and urinalysis in dogs with an insulin-secreting tumor are usually normal. The results of the serum biochemical profile, aside from the blood glucose level, are also usually normal. Hypoalbuminemia, hypophosphatemia, hypokalemia, and increased activity in alkaline phosphatase and alanine aminotransferase have been reported (Leifer et al, 1986), but these findings are considered nonspecific and not helpful in achieving a definitive diagnosis. A correlation has not been established between increased liver enzyme activity and obvious metastasis of pancreatic tumors to the liver.
DIFFERENTIAL DIAGNOSES FOR FASTING HYPOGLYCEMIA
Hypoglycemia is defined as a blood glucose concentration less than 60 mg/dl. Hypoglycemia typically results from excessive glucose utilization by normal (e.g., with hyperinsulinism) or neoplastic cells, impaired hepatic gluconeogenesis and glycogenolysis (e.g., portal shunt), a deficiency in glucose counterregulatory hormones (e.g., cortisol or glucagon deficiency), inadequate dietary intake of glucose and other substrates required for hepatic gluconeogenesis (e.g., anorexia in the neonate or toy breeds), or a combination of these mechanisms (e.g., sepsis) (Table 14-8) (Service, 1995). Iatrogenic hypoglycemia is a common problem with overzealous insulin administration to diabetic animals.
TABLE 14-8 CLASSIFICATION OF FASTING HYPOGLYCEMIA
* IGF-II, Insulin-like growth factor II.
Congenital hepatic disease
Portovascular anomalies are the most common congenital cause of hepatic-induced hypoglycemia. Hypoglycemia develops despite the proper reduction in the circulating insulin concentration because of insufficient hepatic glycogen stores and inadequate hepatocellular function to support gluconeogenesis. Dogs and cats with portovascular anomalies are typically younger than 3 years of age, are thin or cachectic, and may have any of the signs, aside from weight gain, outlined in Table 14-4. Ascites, icterus, hemorrhage, and additional neurologic signs induced by hepatic encephalopathy may also be present. Behavior abnormalities may be enhanced by eating protein and by the administration of drugs (anesthetics, tranquilizers, diuretics). Additional abnormalities suggestive of this disorder that are identified with a CBC, serum biochemistry panel, and urinalysis include microcytosis, hypoalbuminemia, hypocholesterolemia, decreased urea nitrogen, increased total bilirubin, ammonium biurate crystals in the urine, abnormal preprandial and postprandial bile acid concentrations, and small liver size on abdominal radiography or ultrasonography. Confirmatory tests include liver biopsy, angiography, nuclear scintigraphy, and identification of the shunt during abdominal ultrasound scanning or exploratory celiotomy.
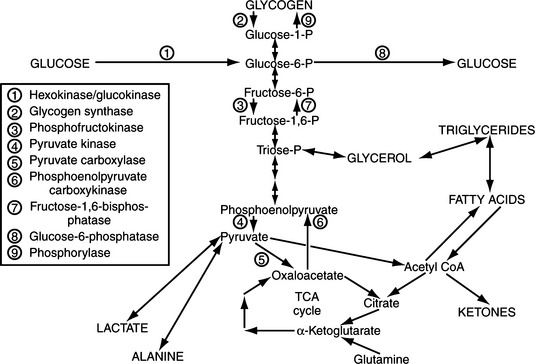
Glycogen storage diseases are rare congenital hepatic disorders in dogs in which hypoglycemia results from an inability to convert glycogen to glucose because of a congenital absolute or relative deficiency of one of the enzymes necessary for the catabolism of glycogen. Type III glycogen storage disease (Cori’s disease), a deficiency in amylo-1,6-glucosidase, has been documented in puppies with clinical signs of massive hepatomegaly (caused by glycogen accumulation), failure to thrive, and muscle weakness (Fig. 14-4) (Hardy, 1989). Glycogen storage disease type Ia (GSD Ia) (von Gierke’s disease) is a deficiency in glucose-6-phosphatase caused by a mutated, defective glucose-6-phosphatase gene. GSD Ia has been documented in the Maltese breed, and a canine model for GSD Ia has been established by cross-breeding Maltese and Beagle dogs (Brix et al, 1995; Kishnani et al, 2001). Affected puppies exhibited tremors, weakness, and neurologic signs when hypoglycemic. Biochemical abnormalities included fasting hypoglycemia, hyperlactacidemia, hypercholesterolemia, hypertriglyceridemia, and hyperuricemia. Affected puppies developed progressive hepatomegaly characterized histologically by diffuse, marked hepatocellular vacuolation. One screening test for glycogen storage disease is the glucagon tolerance test. Plasma glucose concentrations typically fail to increase after injection of glucagon in dogs with type Ia and type III glycogen storage diseases. Confirmatory tests include histologic evaluation of hepatic biopsies and specific hepatic enzyme assays that are rarely performed in veterinary medicine.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


