Chapter 10 Antiviral Therapy
Approximately 80 families and 4000 species of viruses are known to date, and more than 60% of illnesses afflicted humans in developed countries are caused by viruses. However, the development of drugs intended to prevent and treat viral diseases has been frustratingly protracted. Despite the long and intensive search for effective antiviral drugs, very few compounds have clinical applications. Currently, at least 23 antiviral drugs have been approved for use in human medicine; none is approved for use in animals. Unfortunately, unlike the situation with many other anti-infectious drugs, applications for human antiviral drugs in veterinary patients often have been limited because the etiologic agents of viral diseases vary so widely. In recent years, however, given the similarities between feline and human immunodeficiency viruses1 and potentially other viruses, information regarding pathophysiology and drugs with potential efficacy are increasingly applicable to veterinary use. Further, trends have developed toward development of drugs that are broadly effective against viral diseases. The development of species-specific recombinant proteins (e.g., interferons [IFNs]) has increased our knowledge base and promises to improve the therapeutic armamentarium for viral disease afflicting dogs and cats. Although these drugs are discussed in greater depth in other chapters, their support of treatment for antiviral diseases will be addressed here.
In vitro susceptibility testing of viruses requires sophisticated and expensive techniques such as cell cultures. In vitro inhibitory testing procedures have not been standardized, and results vary with the assay system, cell type, and viral inoculum. Additionally, results may not correlate with therapeutic efficacy of antiviral drugs.2 The lack of correlation between in vitro testing and clinical efficacy reflects, in part, the requirement of some antiviral drugs for activation (i.e., metabolism of a prodrug, generally by the host).2 Not only is the spectrum of antiviral drugs narrow, but additionally, a drug often targets a specific viral protein (usually a polymerase or transcriptase enzyme) involved in viral nucleic acid synthesis.2 The limited mechanism of action tends to facilitate the development of antiviral resistance, which can occur rapidly, often reflecting substitution of only a single, although critical, amino acid in the target protein. Drugs that simply inhibit single steps in the viral replication cycle are virustatic. Consequently, viral replication is only temporarily halted, although in human medicine, chronic drug therapy may suppress reactivation of the disease caused by the virus and thus prevent clinical signs of disease. Because drugs often inhibit only active replication, viral growth often resumes once therapy is discontinued.
Viral Replication
Viruses are composed of a core genome consisting of either double-stranded or single-stranded DNA or RNA surrounded by a protein shell known as a capsid. Some viruses are further surrounded by a lipoprotein membrane or envelope. Both the capsid and lipoprotein membrane may be antigenic. Viruses cannot replicate independently and must usurp the host’s metabolic machinery to replicate. Therefore viruses are obligate intracellular parasites. The host’s pathways of energy generation, protein synthesis, and DNA or RNA replication provide the virus with the means of viral replication. For some viruses, replication is initiated by viral enzymes.2 DNA viruses include poxvirus, herpesvirus, adenovirus, hepadnavirus, and papillomavirus. RNA viruses include rubella virus, rhabdovirus (rabies), picornavirus, arenaviruses, arboviruses, orthomyxovirus, and paramyxovirus (canine distemper) (Table 10-1).
Cells respond to viral infection in three ways: infection may have no impact on the cell or its function, cellular death may occur (which may preclude subsequent infection), or the cell may be transformed such that host control of cell growth is lost to viral activities. Viral replication occurs in five or six sequential steps (Figure 10-1): cell entry, including host cell attachment, generally through specific receptors, followed by host cell penetration; disassembly or uncoating resulting in release of viral genome; transcription of viral genome (or viral messenger RNA), which is dependent on virus-specified enzyme; translation of regulatory (early) or structural (late) viral proteins; post-translation modifications (including proteolytic cleavage, myristoylation, glycosylation); assembly of virion components; and release of the virus, generally by budding or cell lysis.2 For DNA viruses, viral DNA is transcribed to host mRNA by host cell mRNA polymerase (or, for poxvirus, viral RNA polymerase). Replication of RNA viruses requires virion enzymes to synthesize mRNA. Double stranded RNA (dsRNA) viruses contain RNA molecules that are transcribed into proteins. Two groups of single stranded RNA (ssRNA) viruses exist. The RNA genome of positive-sense ssRNA viruses is directly translated as mRNA by the host. In contrast, the RNA genome of negative sense ssRNA viruses must first be translated to mRNA by viral RNA-dependent RNA polymerase; host ribosomes subsequently translate to the protein.
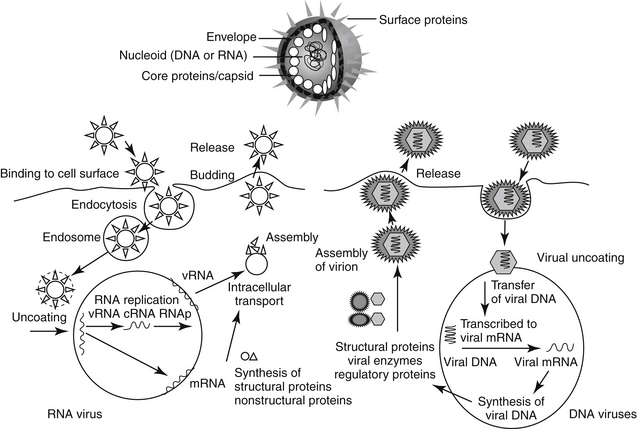
Retrovirues are unique viruses that contain a single strand of RNA that must first be translated, via reverse transcriptase, to a DNA copy of the viral RNA template. The DNA is then incorporated into the host genome (as a provirus), duplicated, and subsequently transcribed into genomic RNA and mRNA for translation into viral proteins.2
In addition to the directed immune response, the host will mount a number of nonspecific protective mechanisms. These include increased body temperature, activity of natural killer cells and phagocytes, and hormones. The role of IFN increasingly is being revealed as a target of pharmacotherapy. Viral infection begins with interaction between the virus and its specific host cell receptor. It is this interaction that initiates the host activation of multiple signal transduction cascades that mount a host defense. Ultimately, these mechanisms cause the nucleus to activate diverse immunoregulatory genes and proteins that cause the intracellular environment to be antagonistic toward viral replication.3 Phosphorylations activate several families of transcription factors. Among the antiviral genes regulated are those encoding interferon (IFN), including α-1 and –β regulators. Several viral cell receptor–initiated events have been identified for their ability to induce activation that ultimately involves IFN. For example, chemokine receptor binding to human immunodeficiency virus (HIV) envelope or glycoproteins, and poxvirus and measles virus binding to T and B cell membrane–bound glycoprotein each initiate such events. The 2–5A pathway is an example of an endogenous antiviral protective system that induces IFN through dependent RNase and 2’-5’ oligoadenylate synthetase (OAS). Viral infection stimulates OAS, which ultimately leads to destruction of both viral and cellular rRNA. Subsequent cell death is similar in appearance to that caused by apoptosis. Viral replication is subsequently prevented.4 Activation of IFN-based and other defense mechanisms also can occur through nonreceptor–mediated mechanisms.3 Not surprisingly, viruses have developed several mechanisms that evade IFN-mediated cell responses. An example includes production of soluble IFN receptors that preclude interaction with normal receptors (e.g., poxviruses and herpesviruses) that would otherwise activate the cascades, downregulation of IFN synthesis (adenovirus), and blocking of phosphorlyations.3
The pathophysiology of infection, including molecular mechanisms, has recently been described for FeLV5 and FIV,1 including the role of selected cytokines in the immune response.6
Antiviral Drugs
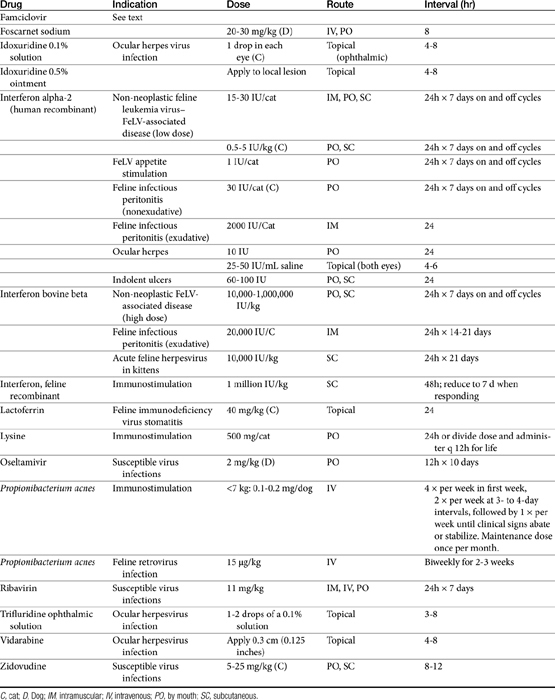
Few antiviral drugs have been studied in animals (“>Tables 10-2, 10-3), and widespread clinical use of antiviral drugs is not common in veterinary medicine. Only a selection of the more promising agents and their purported attributes are briefly discussed; alternatively, when pharmacokinetic information is available, because such information is so limited, it also is provided even if the drug is not an accepted therapy for canine or feline viruses.
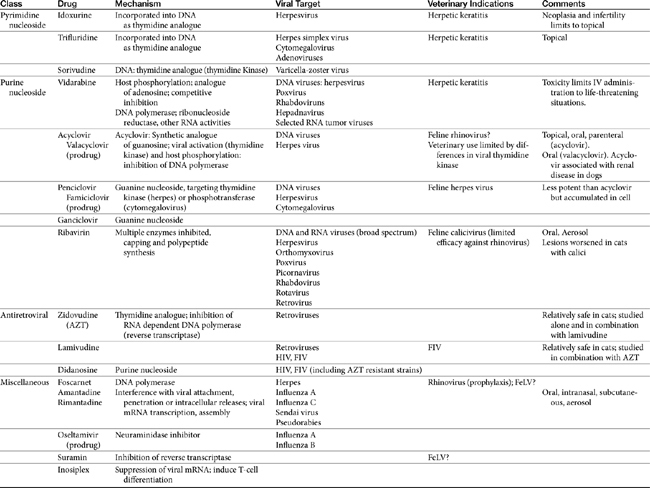
Targets of Antiviral Therapy
Potential targets in the viral life cycle that might be pharmacologically inhibited are expressed during extracellular stages of viral infection (i.e., penetration), intracellular stages (i.e., replication, assembly, and viral release), and dissemination. Those expressed during extracellular stages include specific enzymes whose release is required for skin and mucosal barrier penetration by some viruses, specific cell receptors required for penetration by other viruses, and specific precursor “fusion” proteins that must be activated before cell penetration by some viruses. Antivirals that diminish penetration of host cells by the virus are more viral specific and thus not as inherently toxic as those that prevent viral replication by interfering with viral nucleic acid, DNA, and protein synthesis. Because cell penetration is enhanced by viral-induced immunosuppression, pharmacologic immunomodulation may also help prevent viral penetration.7 Thus these drugs are inherently more useful during the early stages of infection, which are often missed because of the lack of clinical signs. Classes of antivirals that target cell entry include soluble receptor decoys and antireceptor antibodies. Uncoating of the virus can be targeted by ion channel blockers, capsid stabilizers, and fusion protein inhibitors.2
Currently, targets expressed during intracellular stages of viral infection are the most common sites of pharmacologic intervention. Drugs include antivirals as well as a number of other classes of drugs (e.g., immunomodulators). Viral replication depends on macromolecular synthesis (by the host) of viral genome and on genome replication, transcription, and translation. Classes of drugs that inhibit transcription include inhibitors of viral DNA or RNA polymerase, reverse transcriptase, helicase, primase, or integrase. Natural substances capable of inhibiting viral transcription and translation (e.g., IFN) are much more potent than synthetic compounds. Viral replication is targeted by antisense oligonucleotides and ribozymes. Many antiviral drugs are nucleoside or nucleotide analogs, whose chemical structures allow prevention of viral replication by blocking nucleic acid metabolism (Figure 10-2; see also Figure 10-1). However, viral replication is so closely connected to vital functions of the host cell that agents capable of inhibiting viral replication usually injure host cells as well. Although such drugs are more likely to be broad in their antiviral spectrum, most also are potential teratogens, mutagens, and (particularly in humans) carcinogens. Further, they are associated with a variety of other host toxicities, with bone marrow suppression a not uncommon occurrence.7
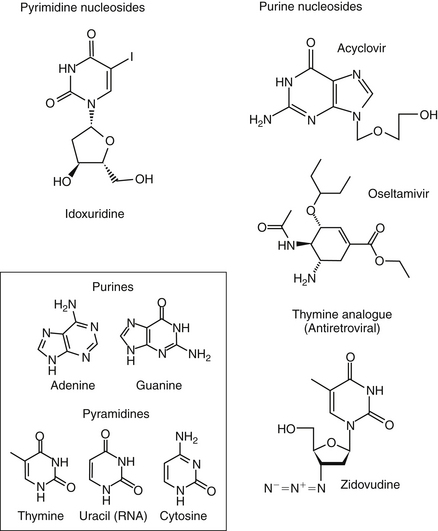
Fewer agents have been developed that block viral translation. Classes of drugs that inhibit viral translation include IFNs, antisense oligonucleotides, and ribozymes. In addition, regulatory proteins might be inhibited. Another category of intracellular targets are specific enzymes, such as RNA or DNA polymerase, or reverse transcriptase of retroviruses, whose expression is required for the maintenance of the viral life cycle. Antiviral agents designed to block expression of these enzymes may have increased selectivity for viruses compared with the host, although their antiviral spectrum frequently is limited. Finally, assembly of synthesized viral macromolecules and release of the assembled virus may be pharmacologically inhibited. For example, IFN-induced inhibition of RNA tumor viruses occurs at assembly, although the mechanism is unknown.7
The final stage of viral infection that might be targeted pharmacologically is release of the new viral progeny from the infected host cell. Viruses can leave cells by causing cellular lysis or budding. With lysis, virus leaves in a sudden burst that kills the host cell. With budding, interaction between the virus and cell receptor induces changes that allow fusion of viral and cellular membranes, thus allowing the progeny to gradually leave the infected cell by budding. An example target for drugs during viral release is the neuraminidase in influenza viruses, a virulence factor that cleaves the sialic acid (neuraminic acid) residues from the glycan portion of cell receptors recognized by viral hemagglutinin.8
Viral infection may be also be pharmacologically inhibited during dissemination, which for some viruses appears to depend primarily on virally-induced host immunosuppression. Thus dissemination is another stage in which modulation of the immune system may help the host overcome viral infection.7 Biological response modifiers emerge with a role in modification of host response, whether it be inhibition of viral-induced immunosuppression or inhibition of an overzealous host response. The biological response modifier most commonly studied for its effect on viral infections and showing the most promise in efficacy has been IFN.
Antiviral-Induced Nephrotoxicity
Among the toxicities caused by antiviral drug is nephrotoxicity. The risk of antiviral drug–induced nephrotoxicity has increased as drugs have become more effective and novel in their action and as combination drug therapy has been implemented in response to increasing viral resistance.9 Acute tubular necrosis has been associated with a number of antiviral drugs (e.g., foscarnet, acyclovir, IFN, and cidofovir). However, variable renal lesions have been ascribed to a number of drugs, reflecting three major mechanisms: transporter defects, apoptosis, and mitochondrial injury. Glomerular disease resulting in proteinuria and, occasionally, the nephrotic syndrome has been mediated by either immune-mediated complexes (IFN) or crystal deposit (foscarnet). Crystalline deposits in the renal tubule (e.g., acyclovir, ganciclovir, and indinavir) can cause intrarenal obstruction. Isolated tubular defects may occur; examples include a Fanconi-like syndrome (cidofovir, tenofovir), distal tubular acidosis (e.g., acyclic nucleotide phosphonates, foscarnet), and nephrogenic diabetes insipidus (NDI: foscarnet).9 A major contributor to toxicity is intratubular cell drug accumulation mediated by ion-transport systems. For example, nephrotoxicity associated with cidofovir and adefovir appears to be facilitated by transport mediated by a transport protein.10 Limited use of antiviral drugs in veterinary medicine probably precludes effective evaluation of the advent of nephrotoxicity in animals with viral infections subsequently treated with antiviral drugs. However, among the clinical signs of toxicosis to accidentally ingested acyclovir in dogs were signs consistent with acute renal failure.11
Antiherpesvirus Drugs
Pyrimidine Nucleosides
Idoxuridine
Idoxuridine (5-iodo-2-deoxyuridine, IDU; Stoxil) was the first of the nucleoside analogues to prove useful in the treatment of viral diseases. Idoxuridine resembles and is substituted for thymidine. After phosphorylation, it is incorporated into both viral and host cell DNA. Altered DNA is susceptible to breakage, resulting in faulty transcription and altered viral proteins. The spectrum of antiviral activity is limited to DNA viruses, particularly members of the herpesvirus group. Resistance to IDU develops rapidly.2 The ability of IDU to cause neoplastic changes, genetic mutation, and infertility limits its use to topical, primarily ophthalmic, infections. IDU is available as an ophthalmic ointment or solution. It is currently approved for use in the treatment of herpes keratitis in humans and has proved useful for the treatment of feline herpetic keratitis. One drop of a 0.1% solution is usually applied to the affected eye every hour; the 0.5% ointment can be applied every 2 hours.12,13 Topical application of IDU to the conjunctiva has been associated with irritation, pain, pruritus, inflammation, and edema of the conjunctiva and punctate areas on the cornea. Resistance of viruses to the drug develops readily both in vitro and in clinical cases.
Trifluridine
Trifluridine (triflurothymidine; TFT; Viroptic) is a halogenated (fluorinated) pyrimidine that is similar and often considered superior to IDU. TFT monophosphate irreversibly inhibits thymidylate synthetase, and TFT triphosphate competitively inhibits DNA polymerase incorporation of thymidine into DNA. Like IDU, it is preferentially incorporated into both viral and host DNA, and late virus-specific DNA transcription is inhibited.2 Trifluridine has in vitro inhibitory effects against herpes simplex virus (types 1 and 2), cytomegalovirus, and selected adenoviruses. Clinical resistance to TFT has been reported. As with IDU, the primary therapeutic indication for TFT is herpetic keratitis. TFT is prepared as a 0.1% ophthalmic solution and is usually applied 6 to 8 times per day. Trifluridine is frequently preferred to IDU for the treatment of human and feline herpetic keratitis in order to prevent toxicities associated with IDU.12,13 Adverse reactions include discomfort on application and palpebral edema.2
Sorivudine
Sorivudine is a pyrimidine nucleoside analog characterized by potency that results in a relative selectivity for varicella-zoster virus (VZV). The drug is initially phosphorylated by viral thymidine kinase and then metabolized to diphosphate by viral thymidylate kinase. As such, sorivudine triphosphate is a competitive inhibitor of viral DNA replication. Unlike acyclovir, however, sorivudine is not incorporated into viral DNA. Inhibitory concentrations of sorivudine are 1000-fold lower for VZV than are those of acyclovir. Cellular uptake in cells infected with herpesvirus is fortyfold greater than in uninfected cells. Clinical resistance has not yet been detected.2
Sorivudine (in humans) is well absorbed after oral administration and is characterized by 98% protein binding. The elimination half-life is 5 to 7 hours, although half-life increases with age. Elimination appears to be urinary, with minimal hepatic metabolism. Side effects are primarily gastrointestinal (nausea, vomiting, and diarrhea). Hepatic enzymes may increase. Long-term administration has caused hepatic neoplasms in rodents. Sorivudine (probably its metabolite) appears to negatively interact with 5-fluorouracil by inhibiting the enzyme responsible for fluorouracil metabolism.2 Sorivudine is available in both oral and intravenous preparations but only as investigational drugs.
Purine Nucleosides
Vidarabine
Vidarabine (Vira-A) was initially investigated for its efficacy as a cancer chemotherapeutic drug. An analog of adenine, vidarabine is phosphorylated by host enzymes and competitively inhibits viral DNA polymerase. It is substituted for adenine into DNA, thus inhibiting viral DNA polymerase. Mammalian DNA is also inhibited, although to a lesser extent. Ribonucleoside reductase, RNA polyadenylation, and transmethylation reactions also are inhibited.2 Vidarabine selectively inhibits DNA viruses, particularly herpesviruses. It is also effective against poxviruses, rhabdoviruses, hepadnaviruses, and selected RNA tumor viruses.2 Until recently, the drug was prepared as an injectable suspension. It is poorly water soluble, however, and must be dissolved in large volumes of fluid before intravenous use. A 3% ophthalmic ointment continues to be available. On intravenous administration, vidarabine is deaminated to hypoxanthine arabinoside which has 10% of the potency of the parent compound, but reaches concentrations that exceed the parent compound by fifteenfold after constant intravenous infusion. The drug is eliminated renally but predominantly as the hypoxanthine metabolite. The elimination half-life of the metabolite is approximately 3.5 hours. Adverse reactions are more likely with intravenous administration and include gastrointestinal upset (vomiting, diarrhea) and central nervous system (CNS) derangements (hallucinations, ataxia, tremors, and painful peripheral neuropathies with long-term use). In addition, vidarabine is probably mutagenic and carcinogenic. Phlebitis, hypokalemia, rash, elevated transaminases, and pancytopenia as well as inappropriate concentrations of antidiuretic hormone have been reported in humans. Systemic use in humans is reserved for life-threatening infections (e.g., herpes encephalitis). Although vidarabine is preferred over IDU for topical therapy of herpetic keratitis, the advent of acyclovir has reduced its use. Vidarabine can be useful for patients that have developed resistance to acyclovir or in combination with acyclovir for life-threatening infections.2 Literature regarding its use in the cat is limited, but topical administration of the 3% ointment appears to be well tolerated in cats.12,13
Acyclovir and Valacyclovir
Acyclovir is an acyclic synthetic purine nucleoside analog that substitutes for guanosine in DNA synthesis. Valacyclovir is an L-valyl ester prodrug of acyclovir. Efficacy of acyclovir depends on activation of the drug to its monophosphate derivative by viral thymidine kinase. Subsequent phosphorylation to the diphosphate and then triphosphate form is mediated selectively by cells infected with herpesvirus. The formation of acyclovir-GTP results in the inhibition of viral DNA polymerase and incorporation of acyclovir-GTP into viral DNA, which terminates viral DNA synthesis. The drug has a greater affinity for viral (versus host) thymidine synthetase. Antiviral activity of acyclovir is limited essentially to herpesviruses. The in vitro activity of acyclovir is 100 times that of vidarabine and 10 times that of IDU. Viral resistance to acyclovir results from mutation to strains that are characterized by a reduction in viral thymidine kinase (the most common mechanism), altered substrate specificity, or altered viral DNA polymerase.2
Acyclovir is available in topical, oral (capsule), and parenteral (powder to be reconstituted) preparations. The bioavailability of the oral preparations (in humans) is poor (10% to 30%) and decreases with increasing doses.2 In contrast, valacyclovir, which is rapidly and completely converted to acyclovir, increases bioavailability of acyclovir to 50% (in humans). Acyclovir distributes to all body fluids, including cerebrospinal fluid. It is eliminated primarily unchanged by glomerular filtration and tubular secretion and accumulates in patients with renal failure. The elimination half-life in adults with normal renal function is 1.5 to 6 hours; this can increase to 20 hours in anuric patients.
Toxicity of acyclovir, regardless of the preparation, is limited. Oral administration (of both acyclovir and valacyclovir) is associated with gastrointestinal upset. Intravenous administration may cause renal insufficiency and (rarely) CNS side effects. Cats experimentally infected with feline herpesvirus type 1 (FHV-1) that received 60 mg/kg daily of valacyclovir orally became ill within 6 to 9 days of therapy, necessitating termination of the study in 12 days. White blood cell counts declined, yet no difference was found in viral pathology in treated versus untreated cats, leading the authors to assess valacylovir as an unlikely option for treatment of FHV-1.14 Renal dysfunction is reversible and may reflect concentration in urine to the point that crystallization occurs2 (see previous discussion). Rapid infusion, dehydration, and inappropriate urine flow increase the risk of renal damage. Phlebitis also may accompany intravenous administration. A retrospective review (January 1995 through March 2000) of acyclovir toxicoses in dogs (n = 105) following accidental ingestion reported by The American Society for Prevention of Cruelty to Animals National Animal Poison Control Center found clinical signs developing in 6 of 10 dogs within 3 hours of ingestion of doses ranging from 40 to 2195 mg/kg. 11 The most common clinical signs were vomiting, diarrhea, anorexia, and lethargy, with polyuria and polydipsia reported in only 1 dog. Treatment included standard decontamination procedures, (i.e., induction of emesis, administration of activated charcoal), diuresis, and supportive care.
Veterinary use of acyclovir may be limited, probably because of differences among infecting viruses in viral thymidine kinase for acyclovir. In addition, antiviral resistance is increasing. Acyclovir is unable to eliminate latent infections. It is available as an ophthalmic ointment, a topical ointment and cream, an intravenous preparation, and various oral formulations.
Penciclovir and Famciclovir
Like acyclovir, penciclovir is an acyclic guanine nucleoside. Its spectrum of activity (herpes simplex virus and VZV) is also similar to that of acyclovir. Penciclovir (up to 77% bioavailable) is formed from its prodrug famciclovir. Penciclovir is a hundredfold less potent than acyclovir but is accumulated to higher concentrations than is acyclovir in infected cells.2 Plasma elimination half-life in humans is approximately 2 hours, and elimination is renal. Although, like acyclovir, the drug is well tolerated orally, chronic administration appears to be tumorigenic, causing testicular toxicity in animals.
The pharmacokinetics and safety of penciclovir resulting from oral administration of famciclovir have been reported in cats (n = 8) at 62.5 mg (half of a 125-mg tablet; 9 to 18 mg/kg) orally after single and multiple dosing (every 8 or 12 hours; 4 cats per group) for 3 days.15 The maximum drug (Cmax; ng/mL) concentration following a single dose was 330 ± 120, and the elimination (disappearance) half-life was 3.1 ± 0.9 hr. Using a multiple-dosing, 12-hour dosing regimen, the Cmax of penciclovir (ng/mL) did not change significantly from single dosing (multiple 12-hour dosing Cmax [ng/mL] of 330 ± 180 at a Tmax of 5 hours) but did with 8-hour dosing (multiple 8-hour dosing Cmax of 680 ± 290) µg/mL. This increase is practically expected with a 3-hour half-life in the face of an 8-hour dosing interval (accumulation ratio of 1.4), but altered clearance cannot be ruled out without intravenous pharmacokinetic studies. The half-life did not change with multiple dosing, although the duration of dosing may not have been enough to allow emergence of drug interactions and the sample size may not have been sufficient to detect a difference. Dose normalization of Cmax and area under the curve (AUC; 24 hours) yielded no significant differences between the two dosing intervals; again, limitations in sample size may have precluded detection of differences. Adverse effects may have included decreases in packed cell volume (by 27%) and total protein (by 10%). However, these changes may have reflected frequent blood draws during the study. White blood cell counts increased (neutrophils and monocytes) by 54% after the last dose. Although the cats tolerated dosing well, the target concentration suggested for treatment of FHV-1 is 3500 ng/m (based on the review of Thomasy et al.15), which was approximately tenfold higher than the Cmax achieved in this study. Further studies must verify the safety of the drug at doses necessary to achieve this concentration.
Ganciclovir
Ganciclovir is structurally similar to acyclovir, with the addition of a hydroxymethyl group on the acyclic side chain. Its spectrum includes herpesvirus, with particular efficacy against cytomegalovirus; effective concentrations are tenfold to a hundredfold lower than the concentrations effective against other herpesviruses.2
Unfortunately, similar concentrations are inhibitory to bone marrow progenitor cells. As with other guanine nucleosides, ganciclovir inhibits viral DNA synthesis after monophosphorylation mediated by viral thymidine kinase (herpes) or phosphotransferase (cytomegalovirus). Diphosphates and triphosphates of ganciclovir are formed by cellular enzymes. The triphosphate competitively inhibits deoxyguanosine triphosphate incorporation into both viral and host DNA, with preferential inhibition of viral over host DNA polymerase. Intracellular concentrations (which exceed acyclovir concentrations by at least tenfold) decline much more slowly than those of acyclovir, resulting in a cellular elimination half-life of approximately 24 hours. Hence the drug is given once daily in humans.2 Resistance most commonly reflects point mutations or deletions in viral DNA, resulting in reduced formation of viral phosphotransferase.
Ganciclovir is poorly bioavailable in humans (9%, with food). More than 90% of the absorbed drug is eliminated renally, with an (plasma as opposed to cell) elimination half-life of 2 hours. Elimination half-life increases proportionately with creatinine clearance. The primary adverse effect is myelosuppression, with neutropenia occurring in up to 40% and thrombocytopenia in 5% to 20% of patients. Myelosuppression more commonly occurs with intravenous administration and is generally reversible by 1 week after discontinuation of therapy, but it can be persistent and fatal. Treatment with granulocyte colony-stimulating factor may minimize neutropenia.2 The risk of myelosuppression is increased when ganciclovir is combined with other cytotoxic drugs. Side effects in the CNS also are frequent, occurring in up to 15% of human patients. Clinical signs include convulsions and coma. Other adverse effects include infusion-related phlebitis, azotemia, anemia, fever, hepatic dysfunction, nausea, vomiting, and eosinophilia. Therapeutic use of ganciclovir includes cytomegalovirus retinitis, particularly in humans with acquired immune-deficiency syndrome–induced immunodeficiency. Ganciclovir is also used for treatment of any infection or prevention of infection (particularly in transplant recipients) associated with cytomegalovirus.
Ribavirin
Ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide; Virazole) is a purine nucleoside analog that is activated by viral phosphorylation and subsequently prevents the formation of mRNA and translation of viral genome.12,13 The action of ribavirin involves specific inhibition of virus-associated enzymes, inhibition of the capping of viral mRNA, and inhibition of viral polypeptide synthesis. Thus it is effective against both DNA and RNA viruses and is a broad-spectrum antiviral drug. Susceptible viruses include adenoviruses, herpesviruses, orthomyxoviruses, poxviruses, picornaviruses, rhabdoviruses, rotaviruses, and retroviruses. Viral resistance to ribavirin is rare. Ribavirin is well absorbed in humans, widely distributed in the body, and eliminated by both renal and biliary routes as both parent drug and metabolites; it has a plasma half-life of 24 hours in humans. It does not have a wide margin of safety in domestic animals. Toxicity is manifested by anorexia, weight loss, bone marrow depression, anemia, and gastrointestinal disturbances. It has been successfully administered by topical, parenteral, oral, and aerosol routes. Ribavirin is administered as an aerosol to human patients afflicted with respiratory viral infections, thus avoiding the hematopoietic toxicities associated with systemic use of the drug. In the cat, in vitro investigations revealed marked antiviral activity against a strain of calicivirus but little efficacy for rhinotracheitis.12,13 However, Povey16 studied the oral administration of 25 mg/kg every 8 hours for 10 days in cats that had been experimentally infected with calicivirus. Pathologic lesions in infected cats worsened, primarily because of severe thrombocytopenia that presumably was drug induced. Cats also developed liver disease, although clinical signs resolved 1 week after the drug was discontinued.
Miscellaneous Antiherpes Drugs
Foscarnet
Foscarnet (phosphonoformic acid) is an inorganic trisodium salt that interferes directly with herpes viral DNA polymerase. The drug may also be effective in treating retroviral infections owing to similar interference with reverse transcriptase. Direct actions preclude the need for intracellular activation. Foscarnet has a hundredfold greater affinity for viral as opposed to host DNA polymerase-α.2 Point mutations in DNA polymerase are responsible for resistance. Foscarnet is poorly bioavailable after oral administration. The drug is concentrated in bones, resulting in complicated plasma elimination. Elimination (in humans) is bimodal with an initial 4- to 8-hour elimination half-life followed by a 3- to 4-day half-life. It is eliminated primarily by the kidneys, with clearance decreasing proportionately with creatine clearance.
Major side effects in human patients include nephrotoxicity and hypocalcemia, which can become symptomatic. Serum creatinine increases in up to 50% of patients but decreases after therapy is stopped. Acute tubular necrosis, crystalluria, and interstitial nephritis have occurred. Sodium loading before therapy may decrease the risk of renal toxicity. Because foscarnet is highly ionized at physiologic pH, metabolic abnormalities are common in human patients. Calcium and phosphorus may decrease or increase. Decreases in ionized calcium may be sufficient to cause clinical signs consistent with tetany. Other CNS side effects reported in human patients (up to 25%) include tremors, irritability, seizures, and hallucinations. Fever, nausea, vomiting, anemia, leukopenia, and hepatic dysfunction also have been reported. Indications in human patients include cytomegalovirus retinitis and herpes infections that are resistant to acyclovir. The disposition of foscarnet and its precursor, thiophosphonoformate (TPFA), have been studied in normal cats. Whereas foscarnet was only 8% bioavailable, TPFA was 44%; however, only 14% of the drug is converted to foscarnet.16a The half-life of foscarnet was approximately 3 hrs with clearance of 1.88 ml/min/kg, being similar to renal clearance. Foscarnet has been shown to be effective prophylactically in the treatment of feline rhinotracheitis. Its efficacy against retroviruses warrants further investigation for the treatment of FeLV. Currently, foscarnet is given to immunocompromised human patients and is being studied in the cat for treatment of retroviral infections.
Antiretroviral Drugs
Zidovudine
All clinically used (human-approved) antiretroviral agents are 2ʹ,3ʹ– dideoxynucleoside analogs. Zidovudine (AZT; Retrovir) is a thymidine analog. Within the virus-infected cell, the 3ʹ-azido group substitutes for the 3’-hydroxy group of thymidine. The azido group is then converted to a triphosphate form, which is used by retroviral reverse transcriptase and incorporated into DNA transcript.12,17 The 3’ substitution prevents DNA chain elongation and insertion of viral DNA into the host cell’s genome, preventing viral replication. Thus the shared mechanism of action of these drugs is inhibition of RNA-dependent DNA polymerase (reverse transcriptase). This enzyme is responsible for conversion of the viral RNA genome into double-stranded DNA before it is integrated into the cell genome. Because these actions occur early in replication, the drugs tend to be effective for acute infections but relatively ineffective for chronically infected cells.2 Cellular α-DNA polymerases are inhibited only at concentrations a hundredfold greater than those necessary to inhibit reverse transcriptase, thus rendering this drug relatively safe to host cells. Cellular γ-DNA polymerase, however, is inhibited at lower concentrations. Zidovudine is effective against a variety of retroviruses at low concentrations (<0.001 to 0.04 μg/mL. The intracellular elimination time of AZT is 3 to 4 hours.2
Among the toxicities caused by AZT is myeloid suppression. The concentration necessary to suppress (human) myeloid cells is higher than that associated with antiviral activity, but nonetheless is still relatively low at 0.3 to 0.6 μg/mL.2 Although metabolites appear to be void of antiviral toxicity, at least one may contribute to myeloid toxicity. Granulocytopenia and anemia are the major adverse effects of AZT in human patients. The risk of toxicity increases in human patients with low (CD4+) lymphocyte counts, high doses, and prolonged therapy. Idovudinzidovudine may cause Heinz body anemia in cats,21 suggesting that complete blood counts should be performed on cats receiving AZT. Granulocyte colony-stimulating factor is indicated for management of granulocytopenia. CNS side effects are more likely as therapy is begun. Other side effects reported in humans include myopathy (characterized by weakness and pain), neurotoxicities, hepatitis (uncommon), and esophageal ulceration. Resolution of myopathy occurs slowly after drug therapy is discontinued. The risk of myelosuppression is increased by drugs that inhibit glucuronidation or renal excretion. Therapeutic indications for AZT in humans include treatment of HIV infections. Treatment with AZT prolongs survival, decreases the incidence of opportunistic infections, increases measures of immune function, and decreases HIV antigens and RNA. Zidovudine has been combined with didanosine or zalcitabine for more sustained CD4+ lymphocyte response.
The disposition of AZT has been studied in cats. It is rapidly absorbed in cats after intragastric or oral administration. Administration of a single dose of 25 mg/kg in normal cats by either route generates maximum serum concentrations of 28 ± 7 and 29 ± 15 μg/mL, respectively. Bioavailability for the intragastric route is 70 ± 24%, and for the oral route 95 ± 23%. The elimination half-life is approximately 1.5 hours and volume of distribution 0.82 L/kg. Drug concentrations were above the effective concentration 50 (EC50) of 0.19 μg/mL for FIV for at least 24 hours after either intravenous or oral administration.18 The drug appears safe at this dose despite drug concentrations being well above that associated with myeloid suppression of human cells. Side effects in cats at 25 mg/kg (administered intravenously) were limited to transient restlessness, mild anxiety, and hemolysis.18
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


