Some of the anti-HIV drugs on the market have been used to treat naturally FIV- or feline leukemia virus (FeLV)-infected cats, and improvement of clinical signs and prolongation of life can be achieved in some of these cats using antiviral therapy. Furthermore, feline herpesvirus (FHV)-1 infection, and specifically ocular infections, can be treated with systemic or topical antiviral compounds. Attempts to treat feline infectious peritonitis (FIP) with antiviral compounds have not been very successful. In canine viral infections, antiviral chemotherapy plays only a minor role so far. Overall, antiviral drugs that are available and useful for cats and dogs are limited, and few controlled studies have been performed to support their use. This chapter addresses only drugs that are available on the market and that have been used experimentally or in the field against feline or canine viral infections. Further information on these and some other drugs used to treat human infections are found in the Drug Formulary in the Appendix. “True” antivirals are compounds that interfere with one step (or several steps) in the viral replication cycle. Closer scrutiny of the relationship of the virus to the cell reveals several points at which the viral cycle can be interrupted, including adsorption to and penetration of the cell, uncoating of the viral nucleic acid, the various stages of nucleic acid replication, assembly of new viral particles, and release of infectious virions, if the cell is not destroyed (Fig. 2-1). The most common antiretroviral drugs are inhibitors of the retroviral enzyme reverse transcriptase (RT: e.g., nucleoside analogues). Drugs with a broader spectrum inhibit other viral enzymes such as DNA or RNA polymerases and thus interfere with viral genome replication (e.g., acyclovir [ACV], foscarnet [PFA]) or by inhibiting proteinases (e.g., proteinase inhibitors) that are important for the splitting of precursor proteins during viral assembly. Other drugs target the viral entry by binding to specific receptors that the virus uses for adsorption (e.g., bicyclams, a new class that inhibits the CXCR4 receptor, which is important for HIV and FIV entry), by acting as fusion inhibitors preventing the conformational changes of the virus necessary for the fusion process, or by interfering with viral uncoating (e.g., amantadine) (Table 2-1). Currently used inhibitors of the viral replication cycle can be divided into eight classes of compounds, nucleoside analogue RT inhibitors, nonnucleoside analogue RT inhibitors, nucleoside analogue DNA/RNA synthesis inhibitors, nucleotide synthesis inhibitors, receptor homologues/antagonists, neuraminidase inhibitors, ion channel blockers, and peptides (Web Table 2-1). TABLE 2-1 Effects of Antivirals on Stages of Viral Replication Cycle 3TC, Lamivudine; ACV, acyclovir; AMD3100, plerixafor; Ara-A, vidarabine; AZT, zidovudine; ddC, zalcitabine; ddI, didanosine; d4T, stavudine; GCV, ganciclovir; IDU, idoxuridine; IFN-α, interferon-α; IFN-ω, interferon-ω; mRNA, messenger RNA; PCV, penciclovir; PFA, foscarnet; RTCA, ribavirin; TFT, trifluridine; VAZV, valacyclovir. WEB TABLE 2-1 Classes of Antiviral Drugs That Inhibit the Viral Replication Cycle BDV, Borna disease virus; CHV, canine herpesvirus; CPIV, canine parainfluenza virus; FCV, feline calicivirus; FeLV, feline leukemia virus; FHV-1, feline herpesvirus 1; FIPV, feline infectious peritonitis virus; FIV, feline immunodeficiency virus; HPAIV H5N1, highly pathogenic avian influenza H5N1; n.d., not determined. aEBM, evidence-based medicine: AZT inhibits FIV replication in vitro and in vivo43; it reduces plasma viral load, improves the immunologic and clinical status of FIV-infected cats, increases quality of life, and prolongs life expectancy. In placebo-controlled trials, AZT improved stomatitis and increased the CD4/CD8 ratio in naturally FIV-infected cats.61–63 Neurologic abnormalities also tend to respond favorably to treatment with AZT. In some cats with FIV-associated neurologic signs, a marked improvement occurs within the first days of therapy. Pregnancy of FIV-infected queens is a potential indication for AZT treatment if the owner wants the kittens to be delivered, although in utero transmission occurs infrequently in natural FIV infection. As is the case in HIV, evidence exists that FIV can become resistant to nucleoside analogues. AZT-resistant mutants of FIV can arise after only 6 months’ use. A single-point mutation in the FIV gene was identified that can create resistance to AZT.163 In humans, resistance to AZT frequently develops, but the addition of lamivudine (3TC) to a therapeutic protocol can cause AZT-resistant strains to revert to AZT-sensitive strains. A combination of these two drugs might be a promising approach in FIV-infected cats to prevent resistance development. However, in a trial in experimentally infected cats, an AZT/3TC high-dose combination treatment did not show anti-FIV activity in chronically infected cats, but caused severe side effects.4 AZT is effective against FeLV in vitro.180 It has also been shown to be somewhat effective in treating cats experimentally infected with FeLV when treatment is initiated less than three weeks after infection. When treated less than 1 week after challenge, cats were protected from bone marrow infection and persistent viremia.50 In one study, naturally FeLV-infected cats were treated with AZT and high-dose subcutaneous human IFN-α for 6 weeks; however, treatment with AZT or human IFN-α, or both, did not lead to a statistically significant improvement of clinical, laboratory, immunologic, or virologic parameters.66 In general, therapeutic efficacy of AZT in FeLV-infected cats seems to be less promising than in FIV-infected cats. Studies in which FIV-infected cats were treated with AZT for 2 years showed that the drug is well tolerated in most cats. Hematocrit can decline within 3 weeks of initiating treatment to approximately 60% of baseline but afterwards rebounds in most cases, even without discontinuation of treatment. If hematocrit decreases below 20%, discontinuation is recommended, and anemia usually resolves within a few days.62 Neutropenia is less frequent than anemia. Neutropenia can be prevented or treated with filgrastim in FeLV- but not in FIV-infected cats (in which it may lead to increased FIV loads). Other side effects in cats, including vomiting or anorexia, rarely develop. One side effect that is sometimes positively noted by owners is the development of a fuller and shiny hair coat. For further information, see the Drug Formulary in the Appendix. Stavudine (2’,3’-didehydro-2’,3’-dideoxythymidine [d4T]) is a thymidine-based nucleoside analogue that is closely related in mode of action to AZT because both are thymidine analogues. Stampidine, a derivative of d4T, is currently investigated in human medicine in clinical trials but is not yet commercially available. Stampidine still is an experimental drug but has been used to treat cats that are chronically infected with FIV.187 A single oral (PO) bolus of 50 to 100 mg/kg resulted in a decrease in FIV load in peripheral blood mononuclear cells. A 4-week course of 50 to 100 mg/kg was well tolerated, and cumulative doses as high as 8.4 g/kg were given. Further studies are needed to evaluate the safety and toxicity of this drug for cats. d4T is active against FIV in vitro.7,217 Mutants of FIV that are resistant to d4T and cross-resistant to several other antivirals, including AZT, ddI, and PFA, have been detected.217 No in vivo data in FIV-infected or FeLV-infected cats are published. ddI is active against FIV in vitro,45 and in one experimental study, FIV replication in blood was significantly suppressed in cats treated with ddI.216 Antiretroviral drug-induced peripheral neuropathy, for which the pathogenesis is uncertain, has been commonly reported as an adverse effect in HIV-infected human patients. Neuronal morphology, neurobehavioral testing, viral load, and mitochondrial and neurotrophic factor gene expression were tested after ddI treatment of FIV-infected and uninfected animals. FIV infection resulted in delays in withdrawal latency to a noxious stimulus, which were exacerbated by ddI treatment. Epidermal density of nerve endings was reduced after FIV infection, especially when cats were treated with ddI. ddI decreased mitochondrial cytochromec oxidase subunit I gene expression, and the brain-derived neurotrophic factor expression was downregulated by ddI after FIV infection. Thus, ddI treatment during FIV infection resulted in additive pathogenic effects contributing to the development of antiretroviral toxic neuropathy.216 ddI is also active against FeLV in vitro,180 but in vivo efficacy is still unknown. In vitro, antiviral efficacy has been demonstrated against FIV,122 but no in vivo data exist demonstrating its efficacy in FIV-infected cats. A mutant of FIV that is resistant to ddC was selected in cell culture that showed cross resistance to other antiviral compounds (e.g., ddI, PFA).122 ddC is effective against FeLV in vitro71,142,142 and has been used in experimental studies to treat FeLV-infected cats. It has a very short half-life (clearance and half-life values for ddC in cats are 6.5 mL/min/kg and 54.7 min, respectively)142 and therefore was administered in these studies either via intravenous bolus or via controlled-release subcutaneous implants. Controlled-release delivery of ddC inhibited de novo FeLV replication and delayed onset of viremia; however, when therapy was discontinued (after 3 weeks), an equivalent incidence and level of viremia were established rapidly.71 In a study evaluating the prophylactic antiviral activity against FeLV, ddC was administered by continuous intravenous infusion for 28 days. Doses of 22 and 15 mg/kg/hr were extremely toxic, causing death in 8 of 10 cats. A dose of 10 mg/kg/hr caused thrombocytopenia, and only 1 of 10 cats receiving 5 or 10 mg/kg/hr remained FeLV antigen-negative, although onset of viremia was delayed for several weeks.142 3TC is active against FIV in vitro.4,12 Combination of AZT and 3TC had synergistic anti-FIV activities in primary peripheral blood mononuclear cell cultures.4 FIV mutants resistant to 3TC containing a point mutation in the RT gene were selected in vitro and showed cross resistance to AZT.163 One in vivo study was performed in experimentally FIV-infected cats that were treated with a high-dose AZT/3TC combination (100 or 150 mg/kg/day for each drug). The combination protected some cats when the treatment was started before experimental infection. However, AZT/3TC treatment had no anti-FIV activity in chronically infected cats. Severe side effects, which included fever, anorexia, and marked hematologic changes, were observed in some of the cats with such high-dose dual-drug treatment.4 Data on the anti-FeLV activity of 3TC are not available. The pharmacokinetics of 3TC in cats shows considerable similarity to AZT pharmacokinetics in cats and to that of 3TC in humans.82 Thus, in naturally infected cats, 3TC doses similar to AZT doses are probably recommended. Suramin, 1-(3-benzamido-4-methylbenzamido) naphthalene-4,6,8-trisulfonic acid-sym-3’-urea sodium salt, a sulfated naphthylamine and trypan red derivative, is one of the oldest known antimicrobial agents. In 1904, it was demonstrated that trypan red derivatives are effective in trypanosome infection of mice. Suramin still is a well-known antitrypanosomal agent and is still used in the treatment of African trypanosomiasis (see Chapter 72) and river blindness (onchocerciasis). It also inhibits angiogenesis, and interest has been focused on suramin as a therapy for patients with advanced prostate cancer because of its effects on growth factors involved in prostate cancer cell growth. It also exerts an inhibitory effect on the RT activity of several retroviruses and has been used for treating patients with HIV infection; it has, however, only minor clinical value in human medicine. The antiviral action of suramin is based on inhibition of RT by interacting with the template-primer binding site of the enzyme. Although not a nucleoside analogue, it competitively binds to the primer binding side and inhibits the template-primer binding that is necessary for DNA prolongation. Activity of suramin against FIV is unknown, and no studies on the efficacy of suramin against FIV have been conducted. Suramin was used to treat FeLV-infected cats, although only a limited number of cats have been evaluated. In one study, serum viral infectivity ceased transiently in two cats with naturally acquired FeLV infection during suramin treatment but returned to high levels approximately 14 days after treatment was stopped.20 In another study, six anemic FeLV-infected cats received suramin (10 to 20 mg/kg intravenously (IV) as 10% solution over 3 minutes every 7 days for 7 to 9 weeks), and within 4 to 14 days, erythropoiesis improved. However, progenitor cells remained infected, suggesting that suramin can modulate erythroid differentiation without inhibiting progenitor infection; alternatively, it may inhibit binding of viral glycoproteins to membrane receptors of erythrocyte precursor cells in the bone marrow rather than preventing intracellular virus replication.1 Although effective against FeLV, suramin is associated with a significant number of severe side effects, and the lack of studies involving larger numbers of animals limits its use in veterinary medicine. In humans, side effects include nausea and anaphylactic shock as immediate reactions during administration. Later (after 24 hours), peripheral neuritis leading to palmar-plantar hyperesthesia and photophobia, agranulocytosis, and hemolytic anemia can occur. Another major side effect in humans is the destruction of the adrenal cortex, which is described in almost 50% of the treated patients. Albuminuria often occurs with therapeutic dosages not indicating kidney damage but an excretion of an unknown protein, usually with no other pathologic findings. For further information, see the Drug Formulary in the Appendix. ACV is effective against FHV-1 infection132; however, when efficacy of ACV against FHV-1 and human herpesvirus (HSV) is compared in vitro, ACV is about 1000-fold less active against FHV-1 than against HSV and also significantly less active than other antiherpetic drugs.76,112,131,192 ACV was used in several studies in FHV-1-infected cats but with minor efficacy.57,70,70 The main reason for the poor efficacy against FHV-1 (versus HSV) is the degree of phosphorylation by the herpesvirus-specific thymidine kinase; activity of this enzyme is markedly lower in FHV-1 than in HSV.29 Activity of the thymidine kinase is dependent on the activity of the enzyme deoxycytidine kinase; thus, defects in synthesis of deoxycytidine kinase also can influence virus sensitivity against ACV.195 Many animal herpesviruses, including FHV-1 and pseudorabiesvirus, apparently lack the thymidine kinase-associated deoxycytidine kinase activities. In an in vitro study, FHV-1 exhibited a more than 1000-fold increase in sensitivity when the thymidine kinase encoded by herpes simplex virus-1 (HSV-1) was supplied, also proving that the virus-encoded thymidine kinase is an important determinant of the virus susceptibility to nucleoside analogues.76 When ACV is combined with human IFN-α, synergistic antiviral effects are found195 resulting from the different mechanisms of action of the two drugs; ACV inhibits viral DNA polymerase, and IFN-α interacts mainly with translation of viral proteins. The synergy observed also can result from ACV blocking the synthesis of an IFN-α inhibitor produced by the virus. Efficacy of ACV against canine herpesvirus (CHV) infection is unknown. ACV is commonly used as topical drug. Oral and intravenous administration is less frequently recommended. If used topically in eye infections, frequent application (every 4 to 6 hours) is recommended. In cats, ACV should be combined with human IFN-α or feline IFN-ω because these latter drugs can potentially increase the antiviral effect of ACV. ACV has a relatively low toxicity because it is not activated in uninfected cells. When given systemically in higher doses, when maximal solubility of ACV (2.5 mg/mL at 37° C) is exceeded, however, the drug itself (not the triphosphate) can precipitate in the renal tubules, causing obstructive nephropathy if diuresis is inadequate. In these cases, needle-shaped ACV crystals can be detected in the urine sediment. Urinalysis should be performed regularly in long-term ACV treatment. Renal failure is reversible with adequate rehydration. In a toxicity study in healthy dogs, a short high-dose regimen (210 mg/kg/day via constant infusion for 43 hours), which maintained ACV plasma concentrations, was more detrimental to the kidneys than a longer exposure to a lower dose of the drug given intermittently (15 mg/kg via intermittent infusion every 8 hours for 28 days).84 Accidental ingestion of ACV in dogs seems to be a problem as demonstrated in a retrospective study of 105 cases reported to the National Animal Poison Control Center.134,149 The most common signs of toxicity included vomiting, diarrhea, anorexia, and lethargy; polyuria and polydipsia were reported in only one dog. For further information, see the Drug Formulary in the Appendix. In a placebo-controlled experimental study to determine whether orally administered VAZV can be used safely and effectively, cats with FHV-1 infection were treated with high-dose VAZV (60 mg/kg orally [PO]). Cats appeared to be uniquely sensitive to the toxic effects (renal tubular epithelium and hepatocellular necrosis, severe bone marrow suppression), and even high doses appeared not to suppress FHV-1 replication in these acutely infected cats.133 Efficacy of VAZV against CHV also is unknown. Cidofovir is active against FHV-1 in vitro.76,112,112 It was used in an experimental study including 12 cats with experimental FHV-1 infection. In six cats, cidofovir was used topically (1 drop of 0.5% cidofovir in 1% carboxymethylcellulose in both eyes every 12 hours for 10 days); six cats received a placebo. There was a significant difference in clinical scores and in the amount of ocular virus shedding,42 suggesting the efficacy of cidofovir against FHV-1 infection. Its efficacy against CHV infection is unknown. Penciclovir (2-amino-9-[4-hydroxy-3-(hydroxymethyl)butyl]-6,9-dihydro-3H-purin-6-one, [PCV]) is a guanine analogue used for the treatment of various herpesvirus infections. PCV is absorbed poorly when given orally, and it is used mainly as a topical treatment (e.g., against HSV in herpes labialis). Famciclovir is a prodrug of penciclovir with improved oral bioavailability. However, in a pharmacokinetic study following oral famciclovir administration, pharmacokinetic in cats appeared complex within the dosage range studied. Famciclovir dosages of 15 mg/kg, administered PO every 8 hours, did not result in plasma PCV concentrations with satisfying activity against FHV-1.183 PCV is active against FHV-1 in vitro.112,208 In a study, the efficacy of three antiherpetic nucleoside analogues (ACV, PCV, and cidofovir) against FHV-1 was compared in vitro. Whereas ACV showed very poor ability to inhibit FHV-1 replication, both PCV and cidofovir were nearly equally highly effective. When the infectious dose was raised, the activity of PCV was even superior to that of cidofovir.76,77 Its efficacy against CHV is unknown. Ara-A (9-β-D-arabinofuranosyladenine monohydrate, adenine arabinoside), a purine nucleoside, also inhibits DNA synthesis by being incorporated into DNA and inhibiting DNA-synthesizing enzymes. It is effective in vitro against herpesviruses, poxviruses, and retroviruses, but its clinical use in humans has been restricted to treatment of smallpox and HSV keratitis, dermatitis, and encephalitis.50 Ara-A is phosphorylated intracellularly to Ara-A triphosphate that is incorporated into the DNA of virus (and host), where it terminates elongation. It inhibits DNA polymerase of DNA viruses approximately 40 times more than that of the host. Ara-A is active against FHV-1 in vitro132 and is used topically in FHV-1 ocular infections.115,170 Case reports indicate that a beneficial effect can occur in dogs with CHV infection. In one case, Ara-A was given to five littermates (two puppies had died from CHV infection), and all five survived.18 Ara-A also shows activity against FIP-causing feline coronavirus (FCoV) strains in vitro,9 but no data have been found to demonstrate efficacy in vivo against FIP. IDU is active against FHV-1 in vitro112,132 and is used topically in cats with ocular FHV-1 infection.115,170 Experimentally, systemic use in cats was not effective and caused severe toxicity (e.g., gastrointestinal [GI] disorders, bone marrow suppression).168 Therefore, only topical treatment with IDU is recommended in cats with ocular FHV-1 infection. Treatment of systemic CHV infections with IDU was not successful.18 However, there is one case report in which corneal ulcerations associated with a naturally occurring CHV infection resolved with IDU treatment.92 During topical treatment, frequent application (every 4 hours) is important. Prolonged topical use can cause irritation or nonhealing corneal ulcers. TFT is active against FHV-1 in vitro132 and is used topically in FHV-1 ocular infections.115,170 It has better corneal penetration than IDU. In one study it was shown to be the most potent agent against FHV-1 of all investigated drugs (efficacy in decreasing order: TFT > GCV = PCV = cidofovir = IDU = Ara-A > ACV Virus-specific oligomers have been synthesized that interfere with replication of targeted viruses by interfering with the function of specific nucleic acid sequences. An antiviral phosphorodiamidate morpholino oligomer was used to treat kittens during outbreaks of respiratory disease caused by virulent feline calicivirus.161a For further information on this treatment and its efficacy, see Chapter 14. In vitro, PFA has been shown to be active against FIV,45 but in vivo studies have not been performed. As in HIV infection, PFA-resistant FIV strains can develop.45 PFA is also active against FeLV in vitro,174 but no in vivo data exist. In vitro, PFA has been shown to be active against FHV-1,45 but not as active as other antiherpetic drugs,112,192 and no reliable data exist on its anti-FHV-1 efficacy in cats. Efficacy against CHV is unknown. For further information, see the Drug Formulary in the Appendix. RTCA (1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide) is a broad-spectrum triazole nucleoside that has marked in vitro antiviral activity against a variety of DNA and RNA viruses. The strongest antiviral activity is against RNA respiratory viruses and herpesviruses, and RTCA has been effectively used against HIV infection, Lassa fever (a human adenovirus infection), and hantavirus infections.50 Systemic application, however, is limited because of the development of dose-dependent hemolytic anemia in humans. Thus, RTCA is mainly used by the aerosol route to treat only people with respiratory syncytial virus infection. If used as an aerosol, only low concentrations appear in the systemic circulation, and side effects are tolerable.50 RTCA is a nucleoside analogue, but in contrast to other anti-HIV compounds that act primarily to inhibit RT activity by causing premature chain termination during the transcription of DNA from the single-stranded RNA template, RTCA allows DNA synthesis to occur, but inhibits triphosphate synthesis by inhibiting the enzyme inosine monophosphate dehydrogenase (essential for synthesis of nucleotides) and thus inhibiting nucleotide production. In addition, it prevents the formation of viral proteins, probably by interfering with capping of viral messenger RNA (mRNA). In vitro, RTCA antagonizes the action of AZT, probably by feedback inhibition of thymidine kinase so that the AZT is not phosphorylated. RTCA is active against a significant number of feline and canine viruses in vitro, including FIV,165 FeLV,50 FHV-1,144 feline calicivirus (FCV),144 FCoV,9,201 Borna disease virus (BDV),129 and canine parainfluenzavirus (CPIV).144 In vivo, however, therapeutic concentrations are difficult to achieve because of toxicity, and cats are extremely sensitive to side effects. RTCA is highly active against FCV in vitro. In a study investigating its anti-FCV activity in cats, RTCA administered (25 mg/kg PO every 8 hours for 10 days) beginning either 1 or 4 days after aerosol exposure failed to have any beneficial effect on the clinical course of the disease or to reduce viral excretion. In contrast, enhancement of the severity of clinical findings occurred in the treated group.143 Although active against FCoV in vitro, RTCA was not effective in treating cats with FIP. RTCA was administered (16.5 mg/kg, PO, intramuscularly, or intravenously, every 24 hours for 10 to 14 days) to specific-pathogen-free kittens 18 hours after experimental challenge exposure with a FIP-causing virus. All kittens, including RTCA-treated and untreated kittens, succumbed to FIP. Clinical signs of disease were even more severe in the RTCA-treated kittens, and their mean survival times were shortened.198 RTCA also inhibits canine distemper virus (CDV) in vitro,36 but in vivo studies are still missing. Efficacy of RTCA against BDV was investigated using neonatal gerbils. Intracranial inoculation of RTCA reduced viral propagation in the acutely infected brain, resulting in protection from fatal neurologic disorders, and the results suggested that RTCA directly inhibits BDV replication and might be a potential drug for the treatment of BDV infection.93 The usefulness of RTCA in cats with BDV infection, however, is unknown. For further information, see the Drug Formulary in the Appendix. AMD3100 (1,1′-[1,4-phenylenbis(methylene)]-bis(1,4,8,11-tetraazacyclotetradecane) octachloride dehydrate, JM3100), is the prototype compound among the bicyclams. It is not on the market as an anti-HIV drug but is available for stem cell mobilization in humans100 and can be used in FIV-infected cats. Bicyclams are dimeric low-molecular-weight nonpeptidic compounds that bind selectively to the chemokine receptor CXCR4.159 A common feature of HIV and FIV is the use of a chemokine receptor for infection of primary susceptible CD4+ lymphocytes.154,203,203 Chemokine receptors belong to the group of seven transmembrane proteins, in which signal transmission is afforded through rapid influx of calcium into the cell. During early stages of HIV infection, viral isolates tend to use the chemokine receptor CCR5 as a co-receptor for viral entry, whereas in later stages isolates switch to using CXCR4.13 The major receptor for FIV infection is CXCR4,207 but other receptors have also been shown to mediate viral binding. By blocking the chemokine receptors, infection of cells by HIV or FIV can be prevented.73,207 By binding to CXCR4, the bicyclam prevents the interaction of this receptor with other ligands, such as HIV or FIV, thereby inhibiting the entry of these viruses into the cell.33,35,35 The efficacy of AMD3100 against FIV as such and in combination with 9-(2-phosphonylmethoxyethyl)adenine (PMEA, an investigational nucleoside analogue, not on the market) was investigated in 40 naturally FIV-infected, privately owned cats that were treated in a placebo-controlled double-blind clinical trial. Cats were randomly classified into four treatment groups and treated for 6 weeks with AMD3100, PMEA, AMD3100 in combination with PMEA, or placebo. All compounds were administered subcutaneously (SC), AMD3100 at 0.5 mg/kg every 12 hours, and PMEA at 10 mg/kg twice a week. Treatment of FIV-infected cats with AMD3100 caused a significant decrease in the provirus load, but also a statistically significant decrease in serum magnesium levels without clinical consequences. No development of resistance of FIV isolates to AMD3100 was found during the treatment period.169 Thus, the use of AMD3100 might be a viable approach in the treatment of FIV-infected cats. AMD3100 can be used in a dose of 0.5 mg/kg every 12 hours. Magnesium and calcium levels should be monitored regularly during treatment. In veterinary medicine, oseltamivir might be active against highly pathogenic avian influenza (HPAIV) H5N1 in cats. Oseltamivir has shown good antiviral activity against HPAIV H5N1 in vitro,75 as well as in experimentally infected mice and ferrets,46,96 and is recommended for treatment and prophylaxis of HPAIV H5N1 infection in humans. However, treatment was unsuccessful in tigers during an HPAIV H5N1 outbreak in the Sriracha Tiger Zoo in Thailand in 2004.181 Oseltamivir was administered to the tigers at a dose of 75 mg/60 kg twice daily (human dosage) for treatment and prophylaxis, but failed in symptomatic and asymptomatic animals. The treatment failure may have been the result of improper dosage or timing of drug administration; differences in pharmacokinetics and host metabolisms between humans and felids are also possible. Oseltamivir also might be effective in canine influenza (H3N8); however, the disease is generally mild and self-limiting, and vaccine prevention is available (see Canine Infectious Respiratory Disease, Chapter 6, and Influenza Virus Infections, Chapter 23).
Antiviral and Immunomodulatory Chemotherapy
Antivirals
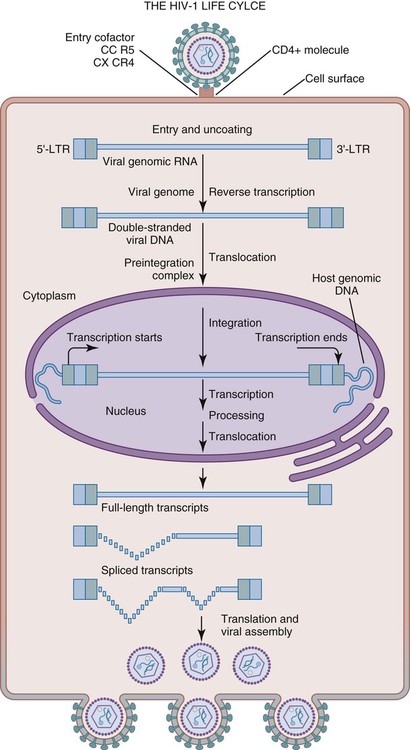
Stage of Virus Replication
Class of Compound
Antiviral Drugs in Veterinary Medicine
Virus attachment
Immunoglobulins
Receptor homologues/antagonists
Immunoglobulin formulationsa
AMD3100
Uncoating
Ion channel blockers
Amantadine
Reverse transcription
Nucleoside analogues
Nonnucleoside/reverse transcriptase inhibitors
AZT, d4T, ddI, ddC, 3TC
Suramin
DNA/RNA synthesis
Nucleoside analogues
Nonnucleotide synthesis inhibitors
ACV, VAZV, Cidofovir, PCV, GCV, Ara-A, IDU, TFT
PFA, RTCA
mRNA translation
Antisense oligonucleotides
Assembly
Interferons
Peptides
Human IFN-α, feline IFN-ω
L-Lysine
Extrusion
Neuraminidase inhibitors
Oseltamivir
Maturation
Glycosylation inhibitors
Proteolytic cleavage inhibitors
Drug
Viral Agent
Efficacy In Vitro
Controlled Study In Vivo
Efficacy In Vivo
Comments
EBM Level (1–4)a
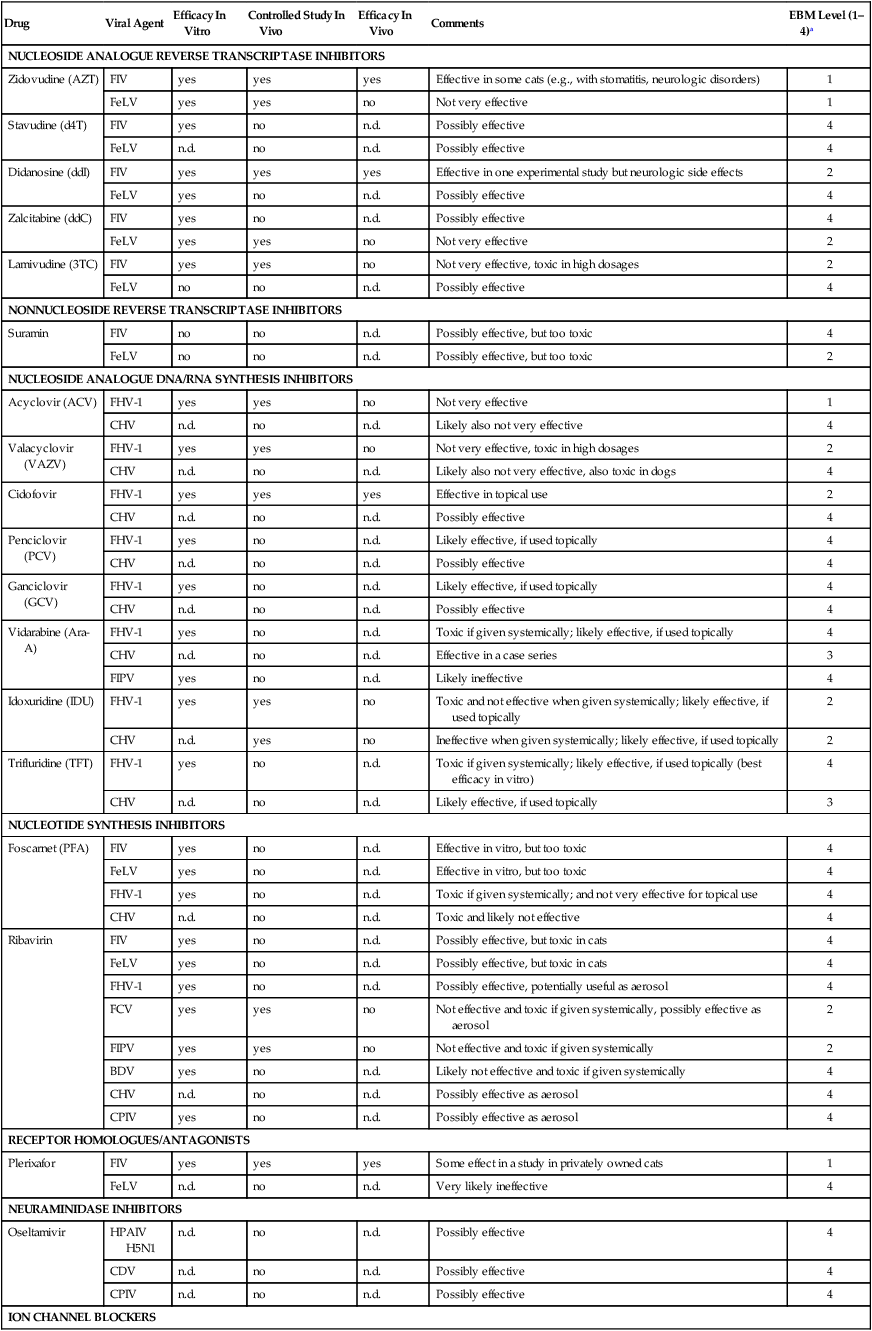
NUCLEOSIDE ANALOGUE REVERSE TRANSCRIPTASE INHIBITORS
Zidovudine (AZT)
FIV
yes
yes
yes
Effective in some cats (e.g., with stomatitis, neurologic disorders)
1
FeLV
yes
yes
no
Not very effective
1
Stavudine (d4T)
FIV
yes
no
n.d.
Possibly effective
4
FeLV
n.d.
no
n.d.
Possibly effective
4
Didanosine (ddI)
FIV
yes
yes
yes
Effective in one experimental study but neurologic side effects
2
FeLV
yes
no
n.d.
Possibly effective
4
Zalcitabine (ddC)
FIV
yes
no
n.d.
Possibly effective
4
FeLV
yes
yes
no
Not very effective
2
Lamivudine (3TC)
FIV
yes
yes
no
Not very effective, toxic in high dosages
2
FeLV
no
no
n.d.
Possibly effective
4
NONNUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS
Suramin
FIV
no
no
n.d.
Possibly effective, but too toxic
4
FeLV
no
no
n.d.
Possibly effective, but too toxic
2
NUCLEOSIDE ANALOGUE DNA/RNA SYNTHESIS INHIBITORS
Acyclovir (ACV)
FHV-1
yes
yes
no
Not very effective
1
CHV
n.d.
no
n.d.
Likely also not very effective
4
Valacyclovir (VAZV)
FHV-1
yes
yes
no
Not very effective, toxic in high dosages
2
CHV
n.d.
no
n.d.
Likely also not very effective, also toxic in dogs
4
Cidofovir
FHV-1
yes
yes
yes
Effective in topical use
2
CHV
n.d.
no
n.d.
Possibly effective
4
Penciclovir (PCV)
FHV-1
yes
no
n.d.
Likely effective, if used topically
4
CHV
n.d.
no
n.d.
Possibly effective
4
Ganciclovir (GCV)
FHV-1
yes
no
n.d.
Likely effective, if used topically
4
CHV
n.d.
no
n.d.
Possibly effective
4
Vidarabine (Ara-A)
FHV-1
yes
no
n.d.
Toxic if given systemically; likely effective, if used topically
4
CHV
n.d.
no
n.d.
Effective in a case series
3
FIPV
yes
no
n.d.
Likely ineffective
4
Idoxuridine (IDU)
FHV-1
yes
yes
no
Toxic and not effective when given systemically; likely effective, if used topically
2
CHV
n.d.
yes
no
Ineffective when given systemically; likely effective, if used topically
2
Trifluridine (TFT)
FHV-1
yes
no
n.d.
Toxic if given systemically; likely effective, if used topically (best efficacy in vitro)
4
CHV
n.d.
no
n.d.
Likely effective, if used topically
3
NUCLEOTIDE SYNTHESIS INHIBITORS
Foscarnet (PFA)
FIV
yes
no
n.d.
Effective in vitro, but too toxic
4
FeLV
yes
no
n.d.
Effective in vitro, but too toxic
4
FHV-1
yes
no
n.d.
Toxic if given systemically; and not very effective for topical use
4
CHV
n.d.
no
n.d.
Toxic and likely not effective
4
Ribavirin
FIV
yes
no
n.d.
Possibly effective, but toxic in cats
4
FeLV
yes
no
n.d.
Possibly effective, but toxic in cats
4
FHV-1
yes
no
n.d.
Possibly effective, potentially useful as aerosol
4
FCV
yes
yes
no
Not effective and toxic if given systemically, possibly effective as aerosol
2
FIPV
yes
yes
no
Not effective and toxic if given systemically
2
BDV
yes
no
n.d.
Likely not effective and toxic if given systemically
4
CHV
n.d.
no
n.d.
Possibly effective as aerosol
4
CPIV
yes
no
n.d.
Possibly effective as aerosol
4
RECEPTOR HOMOLOGUES/ANTAGONISTS
Plerixafor
FIV
yes
yes
yes
Some effect in a study in privately owned cats
1
FeLV
n.d.
no
n.d.
Very likely ineffective
4
NEURAMINIDASE INHIBITORS
Oseltamivir
HPAIV H5N1
n.d.
no
n.d.
Possibly effective
4
CDV
n.d.
no
n.d.
Possibly effective
4
CPIV
n.d.
no
n.d.
Possibly effective
4
ION CHANNEL BLOCKERS
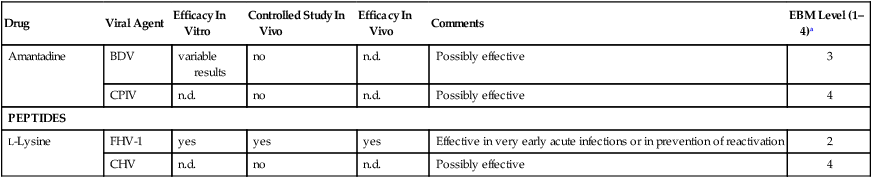
Amantadine
BDV
variable results
no
n.d.
Possibly effective
3
CPIV
n.d.
no
n.d.
Possibly effective
4
PEPTIDES
L-Lysine
FHV-1
yes
yes
yes
Effective in very early acute infections or in prevention of reactivation
2
CHV
n.d.
no
n.d.
Possibly effective
4
Nucleoside Analogue Reverse Transcriptase Inhibitors
Zidovudine
Stampidine and Stavudine
Didanosine
Zalcitabine
Lamivudine
Nonnucleoside Reverse Transcriptase Inhibitors
Suramin
Nucleoside Analogue DNA/RNA Synthesis Inhibitors
Acyclovir
Valacyclovir
Cidofovir
Penciclovir
Vidarabine
Idoxuridine
Trifluridine
 PFA).132 Efficacy against CHV is unknown. However, there is one case report in which corneal ulcerations associated with a naturally occurring CHV-1 infection resolved with TFT treatment.92 Frequent application (every 4 hours) is necessary in topical use.
PFA).132 Efficacy against CHV is unknown. However, there is one case report in which corneal ulcerations associated with a naturally occurring CHV-1 infection resolved with TFT treatment.92 Frequent application (every 4 hours) is necessary in topical use.
Antisense Oligonucleotides
Nucleotide Synthesis Inhibitors
Foscarnet
Ribavirin
Receptor Homologues/Antagonists
Plerixafor
Neuraminidase Inhibitors
Oseltamivir
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Antiviral and Immunomodulatory Chemotherapy
