Chapter 7 Antimicrobial Drugs
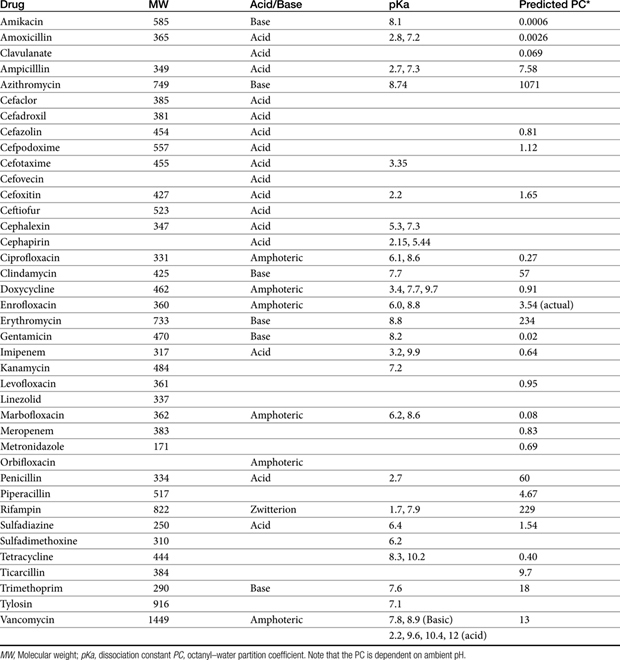
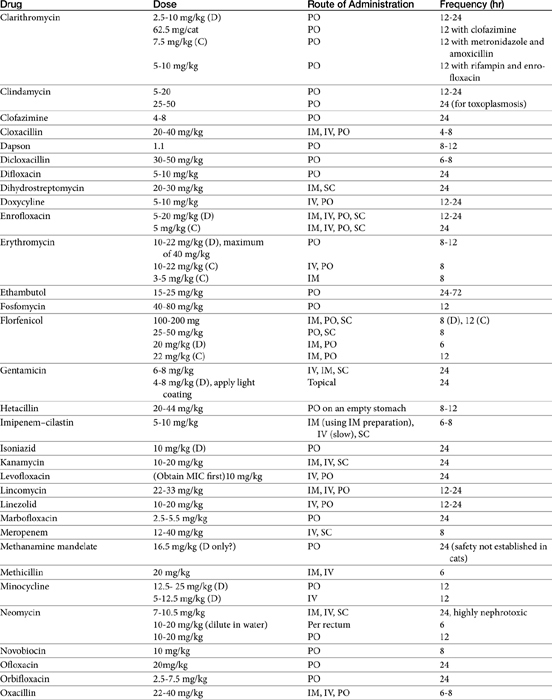
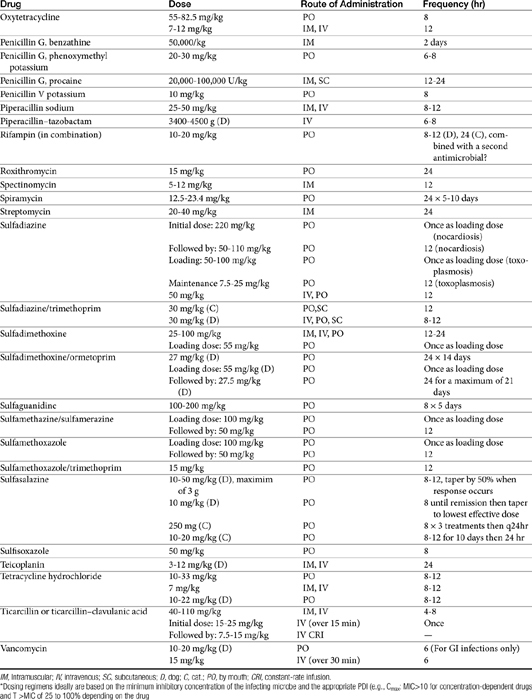
The principles that guide proper antimicrobial selection are discussed in Chapter 6. This chapter focuses on the individual drugs or drug classes and their use to successfully treat bacterial infections. This includes not only resolution of clinical signs but avoidance of resistance. Characteristics discussed for each drug class include structure–activity releationship; the mechanism of antimicrobial action, including whether the drug is time- or concentration- dependent (Table 7-1); the spectrum of antimicrobial activity (Table 7-2), including pharmacodynamics (minimum inhibitory concentrations [MIC] (Tables 7-3 and 7-4) for selected organisms; mechanisms of antimicrobial resistance; clinically relevant aspects of the drug; the disposition of the drug in the patient (as it relates to both safety and efficacy); adverse drug effects; and drug interactions. The breakpoint MICs (the concentration at which an infecting isolate is considered susceptible or resistant to a drug of interest) are delineated in Chapter 6, Table 6-2). Pharmacokinetics were drawn from individual manuscripts, and the Antimicrobial’s Monograph issue of the Journal of Veterinary Pharmacology and Therapeutics2 In addition, Albarellos1 also has provided a review of disposition of selected antimicrobials; these have been included, when appropriate, in Table 7-1. Tissue distribution of antimicrobials is addressed when available; Table 7-5 provides information regarding the relative proportion of tissue versus serum concentrations of drugs, with a focus on body fluids and phagocytic cells. As a reminder (see Chapter 6), drug concentrations measured in tissue homogenates are minimally relevant to concentrations to which microbes are exposed. Data collected by ultrafiltration probes is preferred. However, interstitial fluid is not free of factors that might preclude drug activity (i.e., proteins or ionization; see discussion of cefovecin in cats); as such, dosing errors should be on the side that increase concentrations in tissues. Therapeutic indications are offered when relevant. The dissociation constant of a drug (pKa) and selected information regarding the chemical characteristics of selected drugs or preparation stability are provided for selected drugs in Table 7-6. Doses are indicated in Table 7-7; however, doses ideally should be designed on the basis of intergration of pharmacokinetic (PK) and pharmacodynamic (PD) data (see Chapter 6). Treatment of specific infection is addressed by system in Chapter 8.
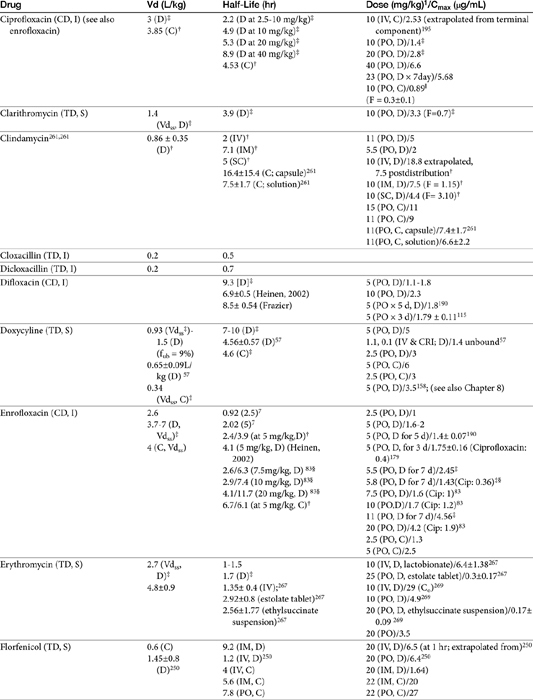
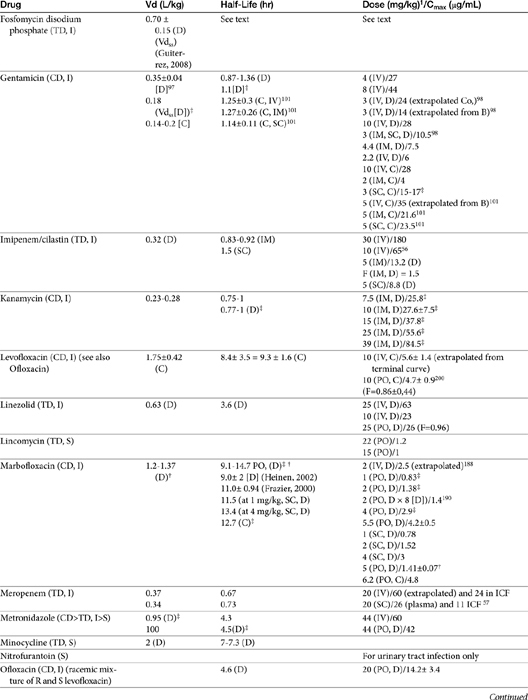
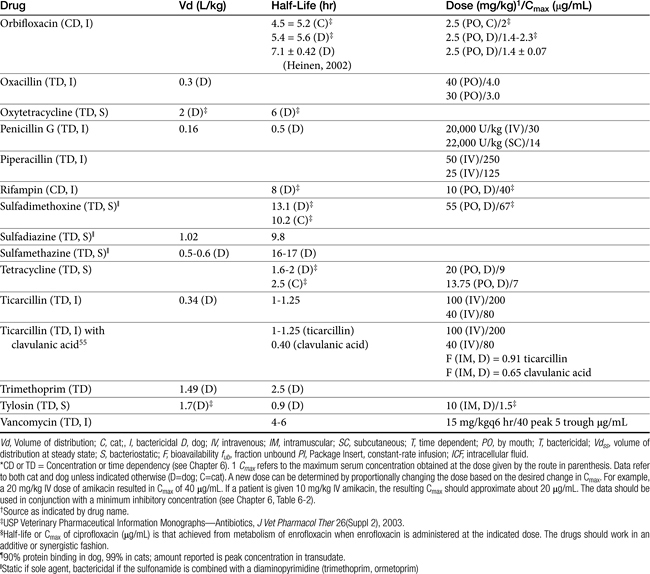
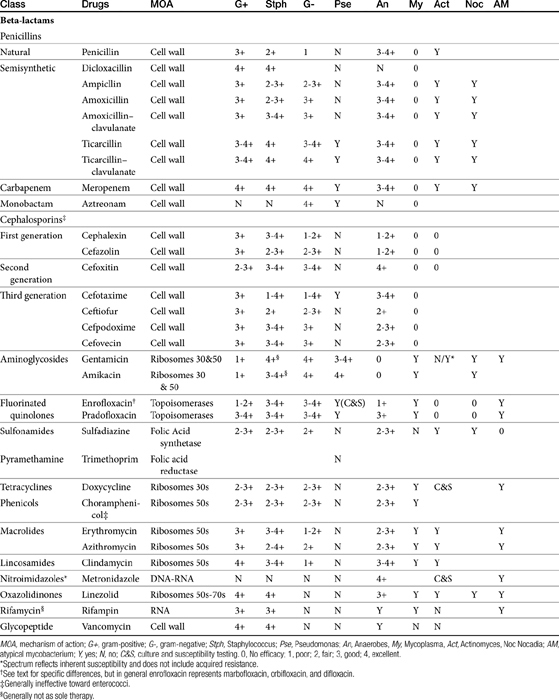
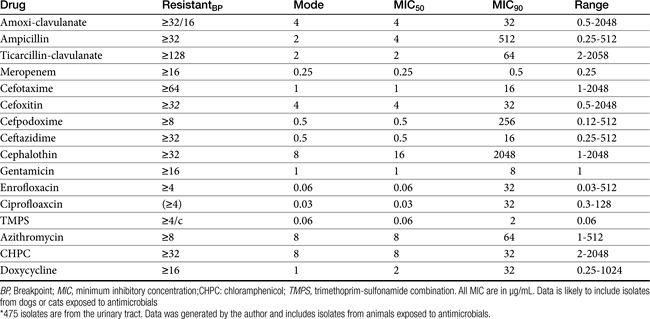
Table 7-4 Susceptibility Data for Selected Drugs and Selected Human Pathogens Associated with Skin and Soft Tissue Infections106
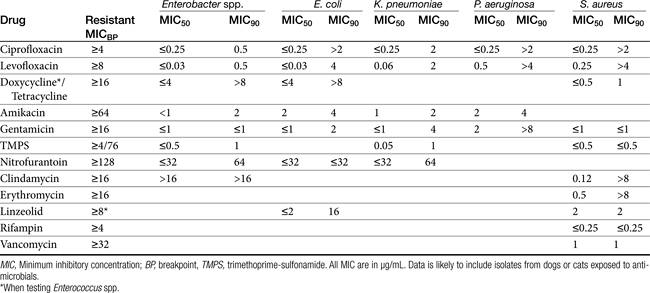
Chapter 6 addressed the importance of integrating PK and PD MIC data when designing a dosing regimen. The PK parameters on which integration is most commonly based are the maximum drug concentration (for both time-dependent and concentration-dependent drugs) and elimination half-life. The latter is particularly important for time-dependent drugs but will also increase area under the curve (AUC), which predicts the efficacy of selected concentration-dependent drugs (e.g., fluoroquinolones; see Table 7-1). Among the sources of PK data to be consulted beyond this chapter are the Antimicrobial Monographs published by the United States Pharmacopiea2 in conjunction with the Journal of Veterinary Pharmacology and Therapeutics. The PD data on which integration is based ideally is the MIC of the isolate cultured from the site of infection in the patient. If not available, the high range of the MIC or the MIC90 might be a reasonable population statistic surrogate indicator of “what is needed” (see Tables 7-3 and 7-4). When available, PD information for canine and feline pathogens (e.g., see Table 7-3) is offered for selected drugs; in addition, relevant information from the human-medicine literature is provided (see Table 7-4). Care should be taken when extrapolating information regarding human pathogens to dogs and cats, although a growing amount of evidence suggests that relative susceptibility of isolates is similar for many drugs (indeed, isolates are likely to be shared), and the data are likely to include both patients that have previously received and not been exposed to antimicrobials. For time-dependent drugs, the relevant PD index (PDI) to be targeted is T > MIC, with a target of at least 50% to 75% of the dosing interval necessary to enhance efficacy, and longer to avoid resistance. An exception can be made for the carbapenems, for which T > MIC of 25% of the dosing interval is sufficient. For concentration-dependent drugs, the relevant PDI is a Cmax/MIC ≥10.3 This ratio should be reached at the site of infection. Alternatively, the AUC/MIC should target 125 to 250. Although as low as 30 has been supported for selected gram-positive drugs, this is particularly true for Streptotoccus pneumoniae, which is an organism that is particularly problematic in humans. This low AUC/MIC may not be relevant to other gram-positive organisms, including other streptococci. Because availability of AUC data is limited, this chapter will focus on Cmax/MIC as the target for concentration-dependent drugs. For PDI for both time- and concentration-dependent drugs, doses should be modified as indicated by drug, host, and microbial factors.
The discussion of antimicrobial drugs is based on their classification by mechanism of action (Figure 7-1; see Table 7-1). The mechanism of action of each drug determines drug efficacy (i.e., bactericidal versus bacteriostatic) and mechanisms of resistance4; influences time- versus concentration-dependence and duration of postantibioitic effect; and, for some drugs, affects safety. Mechanisms of action also influence the selection of combination antimicrobial therapy. For drugs that are approved for use in humans but not animals and for which information regarding use in dogs and cats is not available, PK information in humans will be summarized.
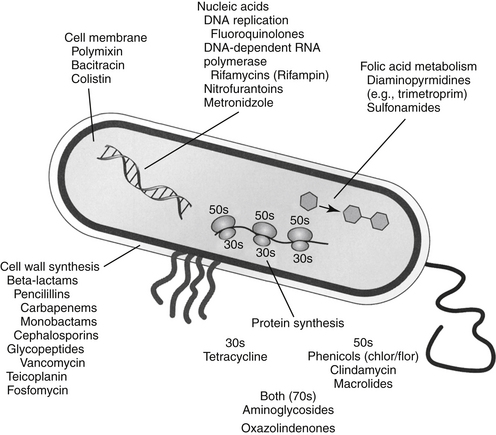
Drugs that Target the Cell Wall
Beta-Lactam Antimicrobials
The broad spectrum, low toxicity, and reasonable cost of beta-lactam antibiotics contribute to their frequent use for treatment of infections. In addition, their effects on cell wall synthesis result in their frequent selection for combination antimicrobial therapy. The beta-lactam antibiotics include the cephalosporins, penicillins (including combination penicillin/beta–lactamase inhibitors), carbepenems, and monobactams (see Table 7-1).
Structure–Activity Relationship
Beta-lactam antibiotics contain a four-member beta-lactam ring as the active site. A second member ring establishes the drug as either a cephalosporin—one carbon larger—or a penicillin (Figure 7-2).5–9 Chemically, the beta-lactams are classified as weak acids (see Table 7-6). They include natural, and semisynthetic drugs (see Table 7-2). Penicillin is a natural drug derived from the molds of the genus Penicillium. Penicillin serves as a base for the semisynthetic aminopenicillins (ampicillin, amoxicillin), the extended-spectrum penicillins (carbenicillin, ticarcillin, piperacillin), the carpabenems (imipenem, meropenem), and the monobactams (aztreonam). Penicillin G is the basis for the definition of the international unit (IU) of penicillin, which is equivalent to 0.6 mg of the international pure crystalline sodium penicillin (1.6 IU/mg). The conversion of USP units varies with the salt, with 1 mg of penicillin G equivelant to the following units: sodium (1500-1750); potassium (1440-1680), and procaine (900-1050).As a group the natural penicillins are unstable and subject to hydrolysis at the beta-lactam ring. Degradation can occur when combined with other solutions. Degradation also occurs for most penicillins exposed to gastric acidity, precluding oral absorption.9
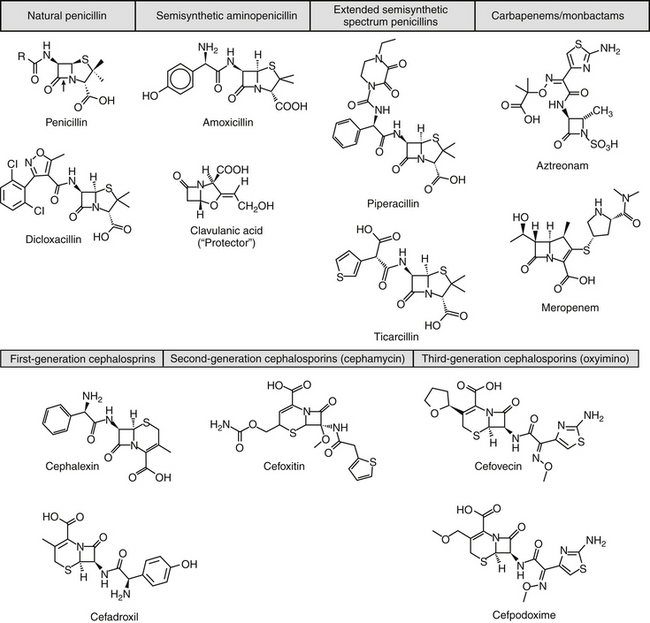
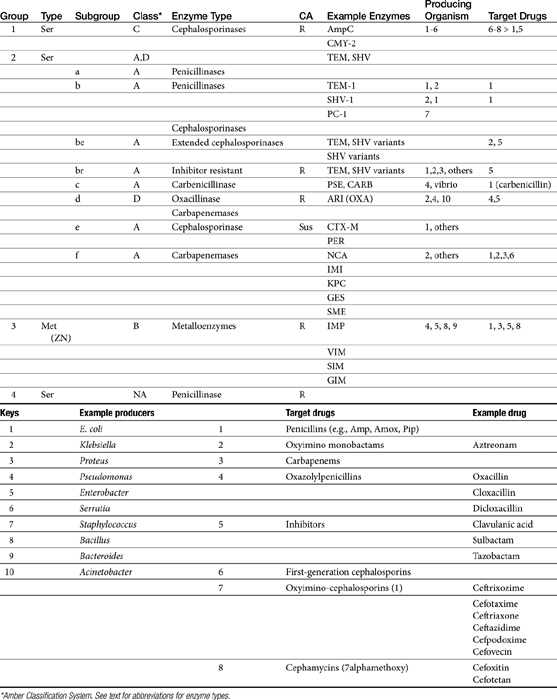
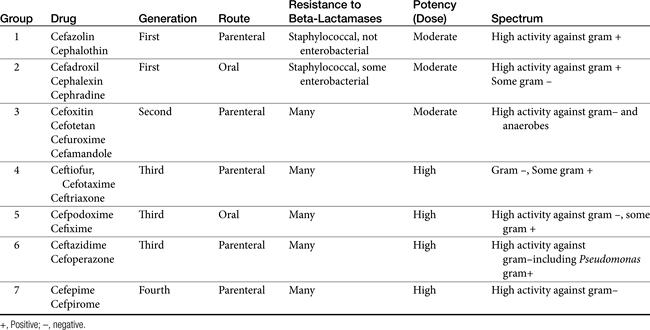
The cephalosporins are derived from a chemical produced by the fungus Cephalosporium acremonium. The six-member ring of the cephalosporins renders them more stable; this increased stability also causes them to be less susceptible to resistance. More than 22 cephalosporins are approved for use in the United States, including the cephamycins (e.g., cefoxitin, cefotetan) and oxyimino-cephalosporins (e.g., ceftazidime, cefotoxime, ceftiofur, cefpodoxime, cefovecin) (see Figure 7-2). The cephalosporins have been variably categorized, with the original “generation” designation being the most widely accepted (Table 7-8).8,10,11 The designations began as an indicator of chronologic approval but have evolved such that each indicates relative resistance to beta-lactamase destruction; the first generation is most and the later generations least susceptible to destruction.10 The advent of extended-spectrum beta-lactamases renders the classification less clear in that these beta-lactams specifically target later-generation drugs. Spectrum and pharmacologic properties of drugs within the generations vary, particularly in the third or later generations. Reclassifying the cephalosporins into groups according to the route of administration, and spectrum has been proposed (see Table 7-8).
Mechanism of Action
The mechanism of action of beta-lactams reflects interference with bacterial cell wall synthesis (Figure 7-3). The bacterial cell wall comprises several layers of a peptidoglycan matrix. The peptidoglycan strands are composed of five repeating disaccharide units of N-acetylglucosamine and N-acetylmuramate; these units are formed by the bacteria in stages. A pentapeptide, which ends with a D-Ala-D-Ala terminus, is attached to each of the repeating units of these disaccharides. The units are joined to form a chain or peptidoglycan strand. The resulting chains are then cross-linked to provide cell wall rigidity. Cross-linking between the D-Ala-D-Ala terminals is catalyzed by transpeptidase enzymes, one of several types of proteins that bind penicillin (referred to as penicillin-binding proteins [PBPs]) located in the cell wall (see Figure 7-3).12 The bacterial substrate for the transpeptidase enzyme is the pentapeptide of the peptidoglycan and, specifically, the terminal amino acids D-Ala-D-Ala. The beta-lactam ring is the functional (active) group of all drugs in this class. It is structurally similar to the D-Ala-D-Ala terminus of the pentapeptide, acting as a substrate and subsequently inhibiting the D-D transpeptidase enzyme (see Figure 7-3). In an actively growing cell, as peptidoglycan precursors increase in response to inhibition of synthesis, autolysins, particularly in gram-positive organisms, contribute to cell wall degradation. Degradation coupled with impaired cell wall synthesis causes the bacterial cell wall to lose rigidity. The cell becomes permeable to the surrounding environment, which, although isotonic to the host, is hypotonic to the organism. Influx of surrounding fluid into the hypertonic bacterial cell results in cytolysis, or osmotic lysis, particularly in gram-negative organisms. Cell wall instability induces the secretion of autolysins, particularly in gram-positive organisms. Because organisms continually break down and rebuild cell walls, the efficacy of the beta-lactam antibiotic ideally is constantly present and, as such, this class of drugs is considered time-dependent (see Chapter 6). However, the duration that the plasma drug concentration (PDC) should be above the MIC varies with the drug, with the desired duration being 50% to 75% of the dosing interval for most drugs. However, T > MIC may be as little as 25% to 50% for carbapenems because they are characterized by more rapid bacterial killing.13 A longer T > MIC is indicated to decrease the risk of resistance.
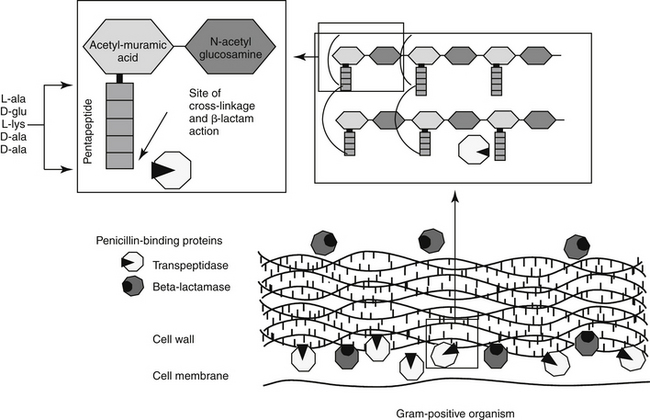
Although all PBPs are able to covalently bind beta-lactam antibiotics, the numbers bound and subsequent activity vary among organisms. Up to nine PBPs are encoded by the genome of Escherichia coli; each PBP generally has subgroups. The diversity of PBPs is responsible, in part, for differences in the spectrum of activity of the beta-lactams. High-molecular-weight PBPs (1, 2, and 3) are essential for microbial growth and survival in Staphylocccocus spp., whereas only PBPs 1 and 2 are critical for Streptococcus spp.; as such, these PBPs are the critical targets of antimicrobial therapy in these organisms.14 In E. coli PBP-2 is essential for cell elongation and PBP-3 for cell division. Because PB-3 appears to complex with PBP-1, -4, and -7 as well as with other proteins,12 effective antimicrobial binding to PBP-3 might have a greater impact than binding to other PBPs in E. coli. The PBP targeted is known for some drugs (e.g, cefpodoxime targets PBP-1a and 1b and PBP-3 (see package insert).
Although beta-lactams are very effective antimicrobials, their unique mechanism of action increases the risk of therapeutic failure in certain conditions, independent of bacterial resistance. Efficacy, particularly toward gram-negatives, is reduced in a hypertonic environment (e.g., the renal intersitium of the normally functioning kidney, an abcess) because osmotic lysis may not occur. Slow growth impairs autolysin activity, which may result in the loss of the bactericidal effect of the beta-lactam antibiotic. Examples might include the combined use of a beta-lactam with a drug that slows growth of the organisms (i.e., a ribosomal inhibitor [see Figure 7-1]), or in a hypoxic environment (e.g., abscess).
Spectrum of Activity
The spectrum of activity of beta-lactam antibiotics varies (see Table 7-2). PD data are available for both the dog and cat for limited drugs (Table 7-9), with selected information provided on human pathogens associated with skin or soft tissue infections (Table 7-10). Penicillin G, a natural antibiotic, is effective against selected gram-positive cocci and both gram-negative and gram-positive anaerobes, but it is beta-lactamase sensitive.5 Selected enterococci are not susceptible to penicillin, and most staphylococci produce beta-lactamases. The gram-negative spectrum of penicillin G is limited but includes Pasteurella multocida. Penicillin V is an orally bioavailable natural penicillin, but its antimicrobial efficacy is reduced.9 Beta-lactamase–resistant isoxazolyl-derivative penicillins include dicloxacillin, cloxacillin, methicillin, and oxacillin. These drugs are effective against gram-positive organisms, including Staphylococcus spp., and gram-negative and anaerobic organisms.
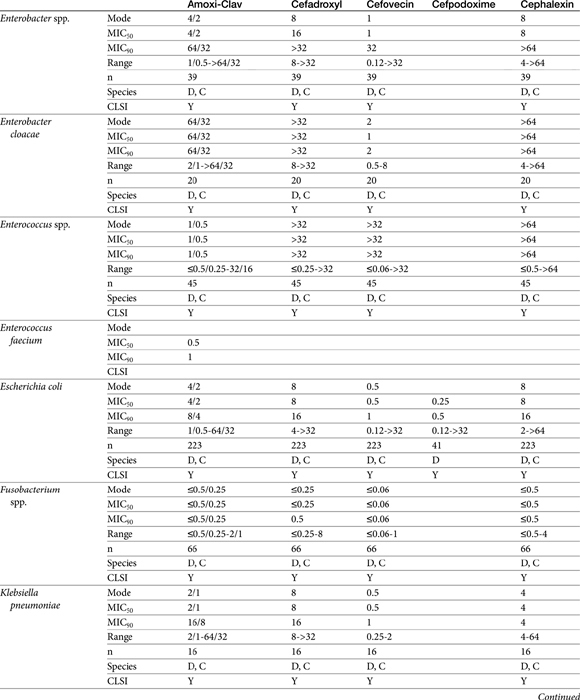
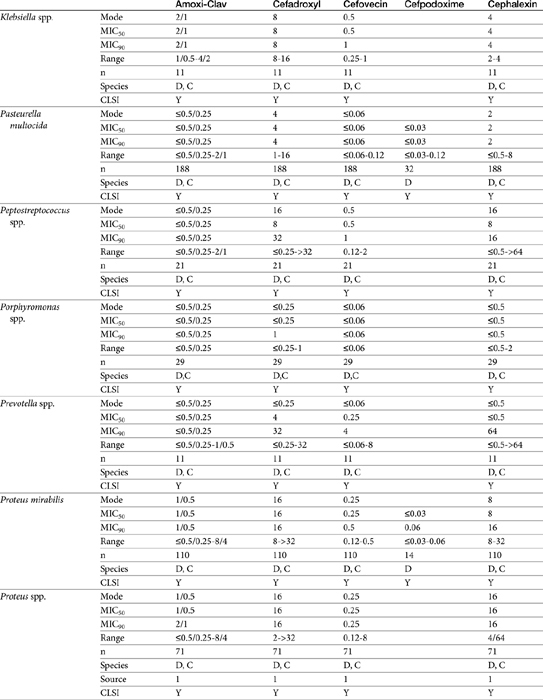
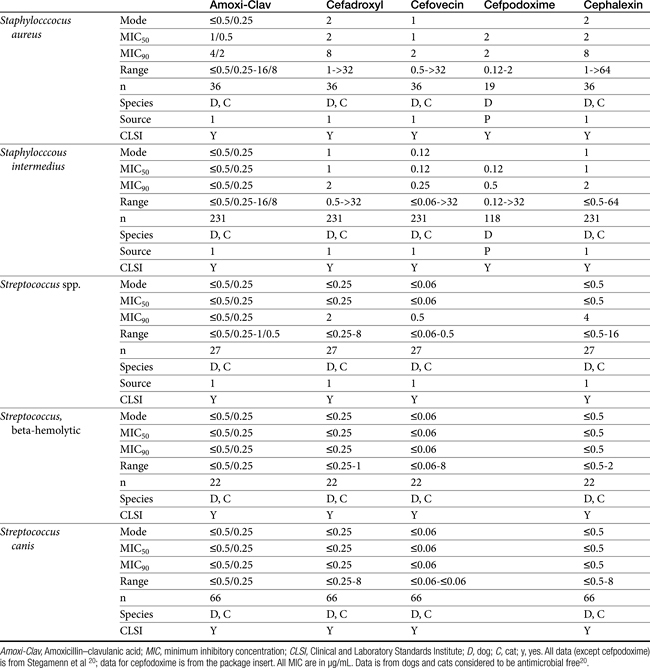
Table 7-10 Susceptibility Data for Selected Human Pathogens Associated with Skin and Soft Tissue Infections160
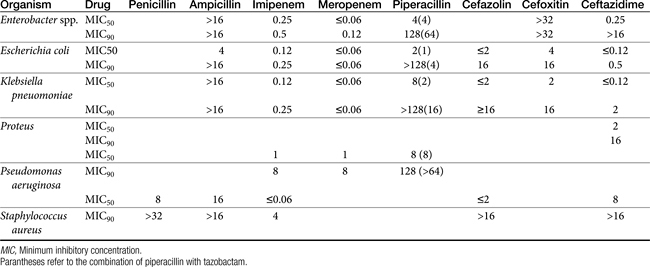
The spectrum of the beta-lactams was expanded with the production of the semisynthetic aminopenicillins. Amoxicillin and ampicillin (aminopenicillins) are considered broad-spectrum drugs; however, this classification has largely been muted by acquired resistance unless combined with clavulanic acid or sulbactam. They target PBP-1a. The anaerobic and gram-positive spectrum of penicillin G is maintained (although the aminopenicillins are slightly less efficacious against anaerobes). The aminopenicillins are generally effective against enterococci, although Enterococcus faecium often expresses resistance. In addition, many gram-negative organisms are added to the spectrum, including E. coli, Pasteurella, some Proteus species, Klebsiella, and selected others (e.g., Salmonella, Shigella). Serratia, Enterobacter, and Pseudomonas are not, however, included in the spectrum of the aminopenicillins. The spectrum of ampicillin is generally similar to that of amoxicillin, and it serves as the model drug for amoxicillin on culture and susceptibility (C&S) testing whereas amoxicillin–clavulanic acid indicates data for ampicillin–sulbactam. However, the potency of ampicillin generally is less than that of amoxicillin against enterococci and Salmonella but greater against Shigella and Enterobacter. The aminopenicillins are less effective compared with the penicillins against Bacteroides fragilis, although efficacy remains good to excellent.9 Like penicillin, the aminopenicillins are beta-lactamase sensitive. Combination with a beta-lactamase protector (e.g., clavulanic acid or sulbactam) improves efficacy and thus broadens the spectrum against susceptible organisms that have acquired resistance through beta-lactamase production. This includes Staphylococcus, E. coli, Klebsiella spp., and some Proteus spp.15 Pseudomonas spp. and other gram-negative organisms remain resistant.7,9 Further modifications led to the extended-spectrum penicillins characterized by a markedly enhanced spectrum, particularly against gram-negative organisms, including Pseudomonas aeruginosa, Serratia, Proteus spp., some Klebsiella spp. Shigella spp., and Enterobacter spp. Examples include the carboxypenicillins carbenicillin and ticarcillin, with ticarcillin having two to four times higher activity toward Pseudomonas spp. than carbenicillin, and the ampicillin-derived ureidopenicillin piperacillin, which has the highest antipseudomonal activity.5,7,16,17 The extended-spectrum penicillins are effective against anaerobic organisms, although they may be less effective than the natural penicillins. They maintain, however, good to excellent activity against B. fragilis.9 The extended-spectrum penicillins are beta-lactamase sensitive; however, a ticarcillin/clavulanic acid combination product is available.
The carbapenems (imipenem and meropenem) and monobactams (aztreonam) represent the most recent members of the beta-lactam penicillins.18 Imipenem targets PBP-1a, -1b, and -2, with its efficacy based on binding to PBP-2 and -1b. It is prepared in combination with cilastatin, which inhibits renal tubular degradation (metabolism by dehydropeptidase-1) of imipenem. As a result, drug half-life may be prolonged (although the clinical relevance of this effect in animals is questionable), and the formation of potentially nephrotoxic metabolites is reduced. Imipenem and meropenem have the broadest antimicrobial spectrums available against bacterial organisms with cell walls, including Pseudomonas spp. Imipenem and meropenem are relatively resistant to beta-lactamase destruction. However, an extended beta-lactamase enzyme has recently been reported, particularly in Klebsiella pneumoniae,emerging as a nosocomial pathogen.19 An advantage of the carbopenems has been their very low MICs (0.05 to 2 μg/mL) for most susceptible organisms. Meropenem is generally similar to imipenem for empirical treatment of serious infections.
Aztreonam is a monobactam (see Figure 7-2), with a high affinity for PBP-3 and lesser affinity for PBP-1a. It is particularly effective against gram-negative aerobes, including Pseudomonas spp. but is ineffective against gram-positive organisms and anaerobes.
The spectrum of the cephalosporins is more diverse than that of the penicillins and is not as easily categorized. Although generalizations regarding the spectrum of activity of each successive generation might be made, variability in efficacy among the drugs within and certainly among generations may result in therapeutic failure if attention is not paid to differences.9 Thus either the package insert or C&S data should be consulted before selecting a cephalosporin, particularly beyond the first generation. In general, cephalosporins are ineffective against enterococci. With each successive generation, the cephalosporins become increasingly more resistant to beta-lactamase destruction, and all generations are generally more resistant as a class than are the penicillins. As such, they are often chosen as empirical first-choice treatment of Staphylococcus spp. Cephalothin (no longer commercially available) has been the drug designated by the Clinical Laboratory Standards Institute (CLSI; previously National Committee for Clinical Laboratory Standards [NCCLS]) as the model indicator for susceptibility for the first-generation cephalosporins. However, it does not represent the class equally. The aerobic spectrum of the first-generation cephalosporins is similar to that of the aminopenicillins,9 although efficacy is more similar to amoxicillin–clavulanic acid combinations. First-generation cephalosporins such as cefazolin, cephalothin, and cephalexin are active (although not equally so) against gram-positive and gram-negative organisms such as E. coli, K. pneumoniae, and Proteus mirabilis. Among the first-generation drugs, cefazolin has better efficacy than cephalexin against gram-negative organisms (e.g., E. coli) but poorer efficacy against Staphylococcus spp.5,9 Efficacy of cephalexin against E. coli is fair to poor. The anaerobic spectrum of the first-generation cephalosporins is fair but less than that of the aminopenicillins.
The second-generation cephalosporins, cefamandole, cefaclor, cefoxitin, and others, are characterized by enhanced activity toward Enterobacter spp., some Proteus spp., E. coli, and Klebsiella spp.5 Cefoxitin has an excellent anaerobic spectrum, particularly against Bacteroides spp.,5,8 although it is less effective than first-generation drugs against gram-positive organisms. Third- (cefotaxime, ceftazidime, cefpodoxime, cefoperazone, cefovecin, and the oxa-beta lactam moxalactam) and fourth-generation (cefepime; not approved in the United States) cephalosporins are generally reserved for serious gram-positive or gram-negative infections (e.g., P. aeruginosa, Enterobacter spp., and Serratia spp.). However, although the efficacy of most of the second-plus generation cephalosporins against E. coli tends to be good to excellent, efficacy against P. aeruginosa9 is variable, and cross-efficacy among members of these generations to any organism should not be assumed. For example, cefoperazone and ceftazidime are among the most effective drugs against P. aeruginosa, although efficacy is less than that of the newer extended-spectrum penicillins. Cefpodoxime and cefovecin are generally effective against E. coli but not effective against Pseudomonas spp. Selected third-generation cephalosporins (e.g., cefotaxime) are effective against anaerobic organisms, whereas others (e.g., ceftazidime, ceftriaxone, and cefpodoxime) are not. Ceftiofur is a third-generation cephalosporin approved for use for canine urinary tract infections. The antimicrobial spectrum of ceftiofur includes gram-positive (Streptococcus spp. and Corynebacterium spp.), gram-negative (Pasteurella, E. coli, and Salmonella spp. but not Pseudomonas spp.), and anaerobic organisms. Ceftiofur is effective against many staphylococcal organisms; however, selection against Staphylococcus spp. should be based on C&S data.9 The first-generation drug cefazolin has been inappropriatly promoted as a generic version of ceftiofur.19a The spectrum of the third-generation drugs cefpodoxime and cefovecin (the former approved in dogs and cats, the latter approved in dogs but used in cats) includes Staphylococcus spp.; cefovecin is also approved for use in the treatment of Streptococcus spp. Both drugs are effective against a variety of gram-negative organisms, including E. coli and Klebsiella, but are not generally effective toward Pseudomonas spp. Stegemann20 has provided PD statistics for a large number of organisms for cefovecin, as well as selected other beta-lactams, some of which are provided in Table 7-9.
Resistance
Bacteria develop resistance to beta-lactams through four major mechanisms: altered or different PBPs such that antibiotic binding does not occur (e.g., staphylococcal organisms and penicillins; enterococcal organisms and cephalosporins); efflux through specific pumps; loss of or changes in porins (especially P. aeruginosa); and inactivation by beta-lactamases. Inactivation by beta-lactamases is most common. Staphylococcus resistance to penicillin appeared as early as 1942; by the late 1960s, more than 80% of medically relevant isolates were resistant to penicillin as a result of beta-lactamase production. Today more than 90% of isolates (human) produce penicillinase.21 The approval of “protected” drugs (i.e., improved the efficacy of selected penicillins), but along with the cephalsoporins, is likely to have contributed to the emergence of altered PBP. This most notorious mechanism of resistance has yielded methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE).
Beta-lactamases
Beta-lactamases are structurally and mechanistically similar to PBPs; indeed, certain PBPs are capable of beta-lactamase activity. Destruction of the beta-lactam (amide) ring reflects its hydrolysis (see Figure 7-2).22 Currently, more than 400 distinct beta-lactamase enzymes are produced by gram-negative, gram-positive, and anaerobic organisms.23,24 Selected examples are listed in Table 7-11. Altlhough clearly a major mechanism of resistance in gram-positive organisms, beta-lactamase production is also the major mechanism by which gram-negative organisms develop resistance.22 Beta-lactamase production occurs as a result of either chromosomal mutations, particularly in gram-positive organisms, or plasmid-mediated resistance in both gram-positive and gram-negative organisms. Beta-lactamases are either constitutive, already present in the cell wall (particularly in gram-negative organisms), or induced by the presence of the antimicrobial drug (in both gram-negative and gram-positive organisms).25 Gram-negative bacteria have the added advantage of secreting beta-lactamases into the periplasmic space such that they are strategically placed before the antibiotic can penetrate the cell wall.9 The beta-lactams are variably susceptible to destruction by beta–lactamases; microbes vary in which enzyme they produce and whether the enzyme is constitutive or inducible (see Table 7-11).
Two major types of beta-lactamases exist: serine-based enzymes and the metallo-beta lactamases. The latter contain a zinc atom that activates water as the destructive site (see Table 7-11).22 Several schemes have been proposed to classify beta-lactamases.The most common scheme is based on the molecular structure (Amber Classification); however, classification according to the target substrate (Bush–Jacoby Classification) may be easier to follow (see Table 7-11). According to the Amber system, Class B enzymes contain the metallo-beta lactamases, but the other three classes are serine-based enzymes. These include classes A (TEM, SHV), C (ampC, targeting cephamycins [cefotetan, cefoxitin]), and D (OXA; targeting protected drugs, such as dicloxacillin, but also protectors such as clavulanic acid). The most prevalent beta-lactamases are class A penicillinases and cephalosporinases, including clinically relevant TEM-1 and 2 or SHV-1 enzymes found in E. coli and K. pneumoniae, and PC-1 enzymes produced by S. aureus.22
TEM-1 and SHV-1 confer high-level resistance to penicillins and first generation cephalosporines but generally do not target the extended-spectrum (selected second- and third- or fourth-generation) cephalosporins or carbapenems. As such, the cephalosporins (cephalosporin C) are generally less impacted by beta-lactamases, particularly those produced by Staphylococcus species.10 However, only a few cephalosporins are stable against anaerobic beta-lactamases. Selected semi-synthetic beta-lactams also are less impacted by beta-lactamases, including most third-generation cephalosporins and imipenem. The semisynthetic dicloxacillin (and oxacillin) is beta-lactamase resistant, with the exception of class D (group 2d). The combination of beta-lactam antibiotics with drugs that inhibit beta-lactamase activity (e.g., clavulanic acid, sulbactam, and tazobactam) increases the potency of the beta-lactam antibiotic, (but not the spectrum) toward susceptible organisms (see Tables 7-2, 7-9 and 7-10). Clavulanic acid irreversibly binds to some but not all beta-lactamases (see Table 7-11).26 Combinations of beta-lactams with beta-lactamase inhibitors are particularly useful against mixed infections and have shown efficacy against selected multiresistant pathogens such as Acinetobacter spp. Aztreonam is generally resistant to beta-lactamase destruction but is susceptible to extended-spectrum beta-lactamases (ESBLs). The presence and diversity of beta-lactamases in canine and feline staphylococcal organisms has been described. As in other species, production is encoded by the blaZ gene, with all four classes of enzymes (A to D) represented genes for classes A, C, and D being plasmid mediated and class B chromosomally mediated.27
Microbes have adapted to each pharmaceutical manipulation intended to combat emergent resistance resulting from beta-lactamase destruction. Third-generation cephalosporins such as cefotaxime and ceftazidime initially were considered indestructible by beta-lactamases.25 However, high-level use has been accompanied by induction and selection for ESBLs in multiple-resistant coliforms,28 particularly in those organisms that produce TEM and SHV enzymes. The genes encoding ESBLs are carried by large plasmids and are able to confer information between bacterial species and strains. The ESBLs are most commonly found in Klebsiella spp. (incidence in North America, 4.4%), E. coli (3.3% to 4.7%), or P. mirabilis (3.1-9.5%), but they also have been detected in other members of the family Enterobacteriaceae and in P. aeruginosa isolates.29–32 The resistant gene codes for mutations in one or more amino acid (serine) substitutions in class A enzymes (TEM or SHV). The resultant change in configuration allows the enzyme to gain access to the drug despite the large oxyimino side chain of these newer-generation drugs.24 Drugs amenable to destruction by ESBL include third-generation cefotaxime, ceftazidime and ceftriaxone, cefpodoxime, and (presumably) cefovecin.22,33 Selected fourth-generation drugs are also susceptible, including cefepime (no longer marketed in the United States).28 Cephamycins (e.g., second-generation cephalosporins cefoxitin, cefotetan) do not appear to be destroyed (although they are destroyed by ampC). Monobactams (i.e., aztreonam) are destroyed. Carbapenems are generally not destroyed by ESBL, nor are beta-lactamase protectors such as clavulanic acid. The use of beta-lactamase protectors appears to reduce the clinical emergence of ESBLs and may reduce the emergence of other resistant pathogens such as Clostridium difficile and vancomycin-resistant enterococci.34 However, the effect (e.g., of the beta-lactamase in the presence of ESBLs) is not always predictable. Decreasesd cephalosporin usage also reduces the advent of ESBLs.
Resistance to ESBLs often is incorporated in plasmids simultaneously conferring resistance to aminoglycosides and sulfonamides.22 Further, ESBL resistance may be associated with non–plasmid-mediated resistance mechanisms such as occurs for quinolones.22 An “inoculum effect” of ESBLs has been described for some drugs and may explain discrepancies among studies: the MIC of the organisms toward cephalosporins increases with a larger (107) compared with smaller (105) inoculum. Because susceptibility may depend on the size of the inoculum at the site of infection,22 ESBLs may not be detected on routine C&S testing.31 Lack of detection of ESBLs may also reflect different levels of activity against the different cephalosporins.
Detection of ESBLs has been based on double disk diffusion techniques. The susceptible cephalosporin (e.g., cefpodoxime, ceftazidime) is incubated with the isolate as the sole drug and in the presence of a beta-lactamase inhibitor; a substantial reduction in the MIC (e.g., fourfold to eightfold) with the combination drugs compared with the cephalosporin by itself indicates an ESBL.35,36 Not all clinical microbiology laboratories have incorporated tests for ESBLs in routine testing procedures.22 The presence of an ESBL should be suspected with organisms resistant to or with high MIC to cefotaxime but susceptible to beta-lactam/beta-lactamase combinations.22 The detection of an isolate with ESBL in a patient with a serious gram-negative bacillary infection should lead to the use of a carbapenem. However, a novel carbapenemase also has been described following isolation in Serratia spp., K. pneumoniae and Enterobacter cloacae.19,22,37 Alternatively, combination of the cephalosporin with clavulanic acid should be considered.
Altered pencillin-binding proteins
The advent of MRSA and multidrug-resistant Enterococcus spp. also has been associated with cephalosporins although it is likely that beta-lactamase inhibitors contributed to its emergence.25 The approval of the cephalosporins in the 1980s was followed by the first MRSA epidemics in the mid-1980s in the United Kingdom; the use of second- and third-generation cephalosporins also was associated with an outbreak of MRSA in Japan.25 In humans, mortality associated with S. aureus bacteremia is 20% to 40%; MRSA has become a leading cause of nosocomial infections in human medicine. The term MRSA was coined in the early 1960s, when these penicillinase-resistant drugs were relatively new, and refers to resistance expressed in vitro to methicillin.21 Although this discussion will focus on MRSA, increasingly, methicillin resistance is being recognized in other species and much of this information is relevant to all methicillin resistant staphylococci (MRS). Over the 30 to 40 years since MRSA was identified, MRSA infections have led to increased mortality and morbidity. The sequelae of MRSA are worse than those associated with beta-lactamase resistance because no alternative therapy remains that is predictably effective.21 In contrast to resistance resulting from penicillinase production which is generally considered low level, infection with MRSA is considered high-level resistance. Further, MRSA isolates are essentially multidrug resistant, that is, expressing resistance to classes other than beta-lactams.
MRSA and methicillin-resistant Staphylococcus pseudintermedius (MRSIG)38 are indicated by the presence of the mecA gene. This gene encodes a mutation in penicillin-binding protein 2a, thus reducing its affinity for the beta-lactam ring, rendering the organism resistant to all beta-lactams. The mecA gene is carried on the staphylococcal chromosomal cassette (SCC); currently five SCCmec have been described.39 Protectors such as clavulanic acid are also unable to bind and thus are ineffective.21 Detection of MRSA or MRSIG (or methicillin-resistance in other staphylococci [MRS]) on C&S testing generally is based on resistance to oxacillin, which is more stable than methicillin in disks used for testing. However, variability in testing methods can profoundly alter results; therefore, cefoxitin might be a more appropriate indicator of multidrug resistance in these organisms.40 Alternative procedures such as polymerase chain reaction or latex agglutination have been used to detect the gene responsible for the formation of penicillin-binding protein 2a (mecA) of MRSA, and other techniques such as pulsed-field gel electrophoresis or multilocus sequence typing identify the specific strain of MRSA (e.g., USA100 or USA300). It is likely that this area of diagnostics will be refined in the next decade and will be applied to other MRS.
Antimicrobials are associated with induction, selection, and propagation of MRSA. The wide use of cephalosporins, in particular, may have contributed significantly to the advent of MRSA. MRSA in human patients has evolved from a hospital-acquired (HA-MRSA; nosocomial) infection (usually USA100) that occurs most commonly in patients whose immune systems are compromised by a community-acquired infection (CA-MRSA), in which otherwise healthy persons are infected, usually in the skin or soft tissue. Crowded conditions, shared items, and poor hygiene increase the risk of CA-MRSA. It is CA-MRSA strain USA300 that appears to be most commonly associated with increased colonization in dogs and cats. In contrast, it is HA-MRSA (USA-100) that is most commonly associated with infections in dogs and cats.40a According to the Center for Disease Control, the incidence of MRSA doubled in human medicine between 1999 and 2006. The impact of MRSA (or other MRS) in veterinary medicine is increasingly problematic, not only because of its impact on the patient but also because of public health considerations. The mec gene has been detected in MRSA organisms infecting dogs,40–42 and MRSA has been associated with infection in dogs.43 However, MRSA also has been found in up to 4% of healthy dogs, with identification complicated by the need for multiple sampling sites (nasal and rectal or perineal). Risk factors for the presence of MRSA in pets or working dogs (e.g., detection and aid dogs) include contact with human hospitals (particularly if patients fed the dogs treats or were licked by the dogs) and children.42 Infections have been isolated to family members and pets in the same household, but this is likely to reflect original transmission from humans to the pet.40–4244 It is likely that colonization is transient in animals. However, healthy pets have been demonstrated to be potential reservoirs for transmission of MRSA to healthy handlers and a potential health risk to immunocompromised patients (humans and presumably other animals in the household). According to the American Veterinary Medical Association, colonization by MRSA is suggested to be an occupational risk for veterinarians, although the frequency of infection associated with MRSA in veterinarians compared with other health professionals has not been documented.
MRSIG45 has a prevalence of 0.58% to 2% in healthy dogs and up to 4% in healthy cats,42,46 with the mec gene present in each canine MRSIG isolate in one study.47 Human colonization with MRSIG is unusual.42 However, MRSIG has been reported as a cause of infection in human patients,42 and transmission from pets with pyoderma to humans has been confirmed.48,49 Although the true public health significance of MRSA and MRSIG (or other multidrug-resistant organisms) in pets is not clear, the fear of infection may be as important as true risk, necessitating proper hygiene and other proactive measures such that human or animal health (including unnecessary euthanasia) is not risked.
The American Veterinary Medical Association offers a website that includes a discussion of MRSA zoonoses, including sources of guidelines that might decrease the risk presented to susceptible humans.49a Among the more important actions that can be taken is establishment of infection control policies and guidelines in each veterinary practice. In general, common sense approaches should prevail (e.g., minimizing intimate contact, maintaining good personal and environmental hygiene practices; see the three D’s approach described in Chapter 6). This includes cleansing of hands of handlers and the paws (or body) of animals that might be exposed to MRSA, including those visiting human health care facilities. Immunocompromised patients are at most risk for MRSA infection acquired from an animal. In such cases the carrier or infected animal should be removed from the environment until successfully treated for MRSA. For dogs with skin infections, cultures are indicated to detect MRSA, particularly in animals for which infection does not resolve. Successful resolution of colonized or infected animals may require both topical (for skin infections) and systemic therapy. Evidence of successful treatment might be based on skin swabs of the ear, nose, and perianal region. Care must be taken to ensure that the laboratory providing culture procedures is well-versed in the diagnosis of MRSA, including speciation of coagulase-positive organisms.
The multidrug resistance associated with MRSA is now evolving toward other (non–beta-lactam) antimicrobials. This reflects, in part, other resistance genes in the gene cassette carrying the mec gene.42 Drugs that are affected include fluorinated quinolones and aminoglycosides. Although newer fluorinated quinolones (e.g., levfloxacin) appear to be more effective than older drugs in vitro, particularly to Staphylococcus, whether this translates to better clinical efficacy is unclear.21 Glycopeptides such as vancomycin are the initial drugs used to treat MRSA in humans, although increasingly vancomycin-resistant Staphylococcus aureus (VRSA) infections have emerged. Linezolid and rifampin are alternative drug choices.
Pharmacokinetics
The beta-lactams are weak acids, which favor oral absorption. Many of the beta-lactam antibiotics, however, are destroyed by the acidity of the gastrointestinal tract and thus cannot be given orally. Penicillin exceptions include penicillin V, dicloxacillin, the aminopenicillins (ampicillin and amoxicillin, including combinations with clavulanic acid), and carbenicillin (indanyl form only; effective concentrations can be achieved only in urine). Lack of stability also may affect the shelf-life of reconstituted products; expiration dates should be adhered to as indicated for the reconstituted product. Orally bioavailable cephalosporins include cephalexin, cefadroxil, and cefpodoxime (third or fourth generation). The oral bioavailability of the cephalosporins also varies among drugs and species.5,9
Many beta-lactams are available as intravenous or parenteral preparations. Absorption from parenteral sites tends to be rapid and complete, with the exception of products that are specifically formulated to allow slow release (e.g., esterified penicillins). Although drug concentrations may persist in circulation longer than non–slow-release preparations (an appealing aspect for time-dependent antimicrobials), older dosing regimens were designed for efficacy against organisms considerably more susceptible to drugs at the time of approval compared with current microorganisms. Thus consideration should be taken to design the dose of these products to compensate for any increase in MIC that may have emerged since the approval of the labeled dose. Selected beta-lactams are highly bound to plasma proteins. Although binding limits distribution into tissues, it also contributes to a long disappearance half-life. Cefpodoxime and, to a greater degree, cefovecin are example of beta-lactams whose long half-life reflects slow release from intravascular protein.20
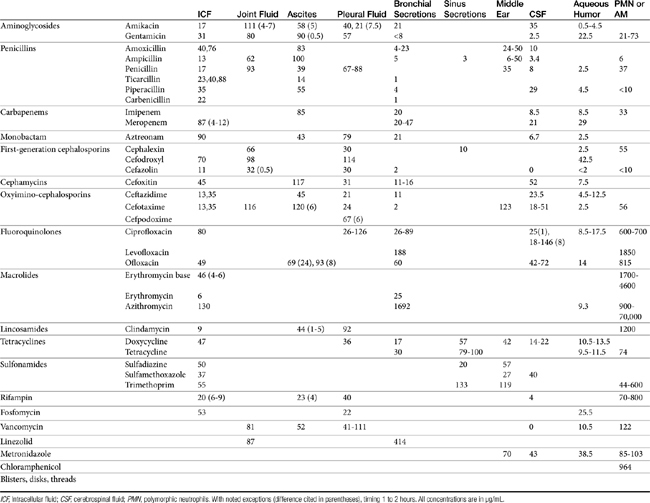
Distribution of beta-lactams is limited to extracellular fluid (volume of distribution [Vd or Vdss] of unbound drug generally ≤0.3 L/kg), but, barring a marked host inflammatory response, adequate concentrations of unbound drug can usually be achieved in the interstitial fluid (the site of most infections) in many tissues (see Table 7-5).5,9 Penicillins and cephalosporins are thus widely distributed throughout most extracellular body fluids, including kidneys, lungs, joints, bone, soft tissues, and bile5,8,11 Interstitial fluid concentrations in normal tissues generally can be predicted by, but are not necessarily equivalent to, the concentration of (unbound) drug in plasma. Comparisons of AUC frequently reveal interstitial fluids to be 30% or less than that in plasma. Among the first-generation cephalosporins, cefodroxil appears to have the better tissue-to-PDC ratio in humans (see Table 7-5). Neither penicillins nor cephalosporins traverse sanctuaries well, including mammary, prostatic, or blood–brain barriers. Imipenem, but generally not antipseudomonal penicillins such as ticarcillin and piperacillin, can reach effective concentrations in the brain. However, first- and second-generation cephalosporins should not be used for central nervous system (CNS) infections because many are destroyed by local enzymes or transported out of the CNS. Beta-lactams in general achieve 25% or less in bronchial secretions compared with PDCs (see Table 7-5).50–52 Inflammation increases the penetration of many beta-lactams. For example, cefuroxime, cefotaxime, ceftriaxone, and ceftazidime can reach therapeutic concentrations when the cerebral spinal fluid (CSF) is inflamed.9 Acute inflammation may also increase beta-lactam penetration of abscesses and pleural, peritoneal, and synovial fluids because of changes in vascular permeability. However, those drugs characterized by high binding to plasma protein will likewise be bound to inflammatory proteins. As response to therapy decreases, resolution of inflammation may decrease distribution. Further, if inflammation does not resolve but progresses, efficacy of beta-lactams is likely to decrease as a result of poor penetrabiltity of lipid tissue. The beta-lactams do not significantly accumulate in phagocytic cells (see Table 7-5). Beta-lactams are concentrated in the urine, enhancing efficacy for cystitis; the clinician must not assume that the high concentration will be achevied in other tissues that also are infected (e.g, nephritis or other urinary tract sites, and even high urinary concentrations may be ineffective in the presence of biofilm (see Chapter 8).
The small Vd that characterizes the unbound beta-lactams contributes to their relatively short half-lives, which often are less than 1 to 4 hours (see Table 7-1). Slow release of highly-protein bound drugs will prolong presence in the plasma. Because beta-lactams in general do not exhibit a long postantibiotic effect, dosing intervals for such drugs may be inconvenient; for critical patients, administering the drug as a constant-rate infusion may be appropriate. The attributes of constant-rate infusion for critical human patients receiving beta-lactams with short half-lives are well recognized and have been demonstrated in animal models.3 The advantages may reflect better steady-state concentrations of drugs in peripheral tissues. Exceptions occur for selected drugs that have a longer half-life, drugs characterized by metabolism to active metabolites, or slowly absorbed or released preparations. The former includes cefpodoxime (4- to 5-hour half-life and 80% to 90% protein bound) and cefovecin (approximate 4- to 5-day half-life and 90% to 99% bound to serum proteins in dogs or cats). Penicillins designed for slow release include slow-release esters (e.g., procaine or benzathine penicillins) or highly protein-bound drugs that may be slowly released from plasma to tissue (e.g., cefovecin). For the latter, generally either absorption or distribution, rather than elimination, half-life is prolonged, resulting in a “flip-flop” model (see Chapter 1). The beta-lactam antibiotics are eliminated, in general, by active tubular secretion in the renal tubules. Clavulanic acid, which is a beta-lactam antibiotic, albeit with poor efficacy by itself, is excreted primarily in the urine of dogs.53 With the exception of hetacillin (no longer available), hepatic metabolism does not play a role in the elimination of the penicillins. Some cephalosporins are eliminated in the urine after deacetylation by the liver, often generate no active metabolites. Examples include cephalothin, cephapirin, cefotaxime, and ceftiofur. Imipenem is degraded to inactive metabolites in the kidney. Reabsorption from the urine is facilitated by an acid urinary pH. Deacetylation of ceftiofur results in an active metabolite; dosing regimens and C&S testing are based on ceftiofur bioactivity.54 Ceftriaxone and cefoperazone are eliminated in the bile in humans and appear to be at least partially eliminated in the bile in dogs.9
Disposition of selected beta-lactam antibiotics
Penicillins
Preparations of penicillin G intended for intramuscular use (e.g., procaine and benzathine) may be prepared as esters, which hydrolyze at variable rates and thus prolong absorption. Procaine penicillin is absorbed for at least 24 hours and benzathine penicillin for approximately 120 hours in some species.9
For the aminopenicillins the oral bioavailability of amoxicillin is greater than that of ampicillin and, unlike ampicillin, is not impaired by the presence of food.5 Clavulanic acid appears to be about 30% to 65% orally bioavailable.15,53,55 The absorption of both amoxicillin and clavulanic acid appears to occur through a saturable process. As with humans, a maximum rate may be reached in dogs at 10 mg/kg and 5 mg/kg, respectively. As the oral dose of amoxicillin reaches 25 mg/kg and clavulanic acid 6.25 mg/kg, amoxicillin may interfere with oral absorption of clavulanic acid. Thus ratios that favor clavulanic acid might be preferred to ensure sufficient absorption.26 Other disposition paramenters of the aminopenicillins are summarized in Table 7-1. The disposition of amoxicillin is such that care should be taken to ensure that underdosing does not occur. This is likely to require administration beyond the label dose (12.5 mg/kg, alone or as clavulanic acid). For treatment of S. pseudintermedius, Stegemann20 has reported an MIC90 of <0.5 μg/mL for amoxicillin–clavulanic acid (see Table 7-9). The MIC50 and MIC90 for amoxicillin–clavulanic acid and E. coli are 2 and 8 μg/mL, respectively. Integration of PK–PD for these organisms indicates that an alternative drug to amoxicillin with or without clavulanic acid might be considered; an exception might occur with UTI because higher drug concentrations will be achieved in the target tissue (urine). However, precaution is also suggested with this approach (see Chapter 8). Note that CLSI has recently re-set breakpoint MIC’s such that many isolates considered susceptible before this change will now be considered resistant.
Carbepenems
Both imipenem and meropenem have been studied in dogs.56,57 Imipenem is minimally protein bound in dogs.56 Peak concentrations (see Table 7-1) occur at 30 minutes for intramuscular and 50 minutes for subcutaneous administration. Extrapolated PDCs after intravenous administration appear to approximate 40 mg/L. The volume of distribution of 0.32 L/kg indicates distribution to extracellular fluid; clearance (CL) is 0.26 L/hr/kg. The elimination half-life varies almost twofold with the route (see Table 7-1). Bioavailability is high after intramuscular or subcutaneous administration.56 In dogs given 5 mg/kg subcutaneously, targeting a 12-hour interval and a T ≥ MIC– (25%) (acceptable for carbapenems), the highest MIC that might be treated is 2 μg/mL. The dose should be increased (approximately 30%) to adjust for ≤70% drug movement from plasma into normal interstitial fluid, particularly if the drug is given subcutaneously.
Meropenem has been studied in dogs after single dose58 and constant-rate infusion.57 As with imipenem, it is minimally (12%) protein bound in dogs. Clearance is 5.6 to 6.5 mL/min/kg. After a dose of 20 mg/kg, mean meropenem (μg/mL) in interstitial fluid (using ultrafiltration techniques) was 24 ± 8 μg/mL. After subcutaneous administration, Cmax (μg/mL) in plasma and interstitial fluid, respectively, were 25 and 11 (ratio = 0.44), and AUCs were 63 and 43 μg ∗ hr/mL, (ratio 0.68) respectively. The better ratio for AUC reflects a longer mean residence time in intracellular fluid (ICF) compared with plasma (2, 4, and 0.9 hours, respectively). Although interstitial fluid concentrations correlated very well with PDC, the doses based on plasma Cmax values might be increased at least 40% when basing dosing on PDC to compensate for differential distribution to extracellular sites of infection. The AUC in interstitial fluid after 20 mg/kg administered intravenously or subcutaneously was 73 μg ∗ hr/mL, and 43 μg ∗ hr/mL, respectively, indicating that intravenous administration might be preferred to subcutaneous administration from a cost standpoint. Note that the time to maximum concentration in interstitial fluid after subcutaneous administration was 3.7 hours (2 hours for intravenous administration), indicating a potential delay in response in the acute situation.58 Based on plasma Cmax after 20 mg/kg administered subcutaneously in dogs, a 12-hour dosing interval, and T > MIC of 25%, the highest MIC that might be treated is 4 μg/mL. If concentrations are used to design the dosing regimen, the highest MIC that could be treated would be 1 μg/mL. Anuric renal failure in humans prolongs the half-life of meropenem fourfold.59
First-generation cephalosporins
Papich et al. described the tissue distribution of cephalexin.59a The ratio of cephalexin Cmax or AUC in plasma versus interstitial fluid were approximately 50% and 57%, respectively. The eliminaton half-life of cephalexin appears to be somewhat route dependent, being almost twice as long as after oral administration (150 minutes) compared with intramuscular or intravenous administration (80 minutes; see Table 7-1). However, Papich et al. reported a much longer half-life of 4.7 + 1 hours in dogs after oral administration of 25 mg/kg.59a Plasma clearance is 2.5 mL/min/kg.60 Bioavailability approximates 60% after either oral or intramuscular administration.60 Oral bioavailability in dogs is affected by the time of day of administration, with Cmax 22% lower in the evening; however, this is more than offset (as a time-dependent drug) by a prolongation of half-life by 50%.61 The oral bioavailability of cephalexin also is affected by pretreatment with metaclopramide, which increases Cmax and AUC, respectively, by 17% and 25%.61 Based on the original half-life reported for cephalexin, targeting T > MIC (50%), the maximum MIC that can be treated using an oral dose of 20 mg/kg is 1 μg/mL. This is equivalent to the MIC50 but less than the MIC90 (2 μg/mL) reported for S. intermedius and cephalexin in dogs.20 A dose of 40 mg/kg is needed for twice-daily dosing, or the interval should be reduced to every 8 hours. Doses would need to be further increased to compensate for differential distribution to tissues or other host or microbial factors. However, if the half-life of 5 hours is used, then twice-daily dosing of cephalexin will result in drug concentration in both plasma and interstitial fluid above the MIC90 for S. intermedius20 for 12 hours or more.59a Note that the MIC50 and MIC90, respectively, for E. coli and cephalexin are 8 and 16,20 indicating that this drug should not be used to treat infections associated with E. coli, including urinary tract infections.
Cephalothin (no longer available in the United States, although it remains the model drug for first-generation cephalosporins at the time of publication) has been studied in dogs after oral administration at 30 mg/kg. Food affects its absorption: Cmax of 45 μg/mL is reduced to 28 μg/mL with food at a Tmax of 1.7 and 2.8 hours, respectively. Elimination half-life is 1.8 and 2.6 hours without and with food, respectively.62
Cefazolin has been studied in two separate groups of canine patients undergoing elective orthopedic procedures. In one study63 clinical canine patients (n = 15) undergoing total hip replacement were administered 22 mg/kg intravenously over 2 minutes at the time of surgical positioning; animals were dosed 2 more times.64 The distribution of the central compartment (Vc; before distribution) was 0.083 ± 0.008 L/kg. The distribution half-life approximated 5 minutes, and the elimination half-life approximated 45 minutes. Tissues from the coxofemoral joint capsule, acetabulum, and femoral cancellous bone were collected from each patient as the site was approached surgically; serum samples were collected at the same general time for each patient. Peak serum concentrations after the first dose were 178 ± μg/mL; tissue (homogenate) concentrations and mean time of collection were as follows: joint capsule, 58 + 5.7 μg/mL at 20 min, acetabulum 157 + 23 at 52 minutes and bone cancellous 227 + 29 at 68 minutes. Peak serum concentrations approximated 178 μg/mL (before distribution) and 119 μg/mL (after distribution). Based on simulations, ideal dosing was suggested to be either 22 mg/kg every 2 hours or 8 mg/kg every hour, to ensure drug concentrations remained above the MIC of Staphylococcus spp. (reported at 2 μg/mL).
Second- and third-generation cephalosporins
Cefuroxime is a second-generation cephalosporin approved for use in humans. Oral administration is in the form of the axetil ester; as a prodrug, desterification occurs before oral absorption. It has been studied both orally and parenterally in Beagles (n=6) as part of a toxicity study.64a Intravenous doses up to 500 mg/kg every 24 hours were well tolerated for 1 week. Jung65 compared cefuroxime in serum to that in cortical tissues in dogs. At approximately 1.25 hours, after 10 and 20 mg/kg administered intravenously, serum concentrations were 12.5 and 28.7 μg/mL, respectively. The elimination half-life was 2.9 hours. Spurling66 reported limited PDCs after oral administration in Beagles. Concentrations (μg/mL) after oral administration of the axetil form at 100 or 400 mg/kg were approximately 28.7 ± 5, and 77 ± 17, respectively.66,67
After a dose of 50 mg/kg ceftriaxone (third generation) was given to apparently healthy dogs, clearance was 3.61 ± 0.8 mL/kg/hr; Tmax occurred at 30 minutes compared with 90 minutes after subcutaneous administration. Pain occurred at the injection site after both intramuscular and subcutaneous administration, whereas intravenous administration was not associated with any adversity.68
Based on studies in dogs after an intravenous dose of 14 mg/kg, cefepime was distributed to a volume of 0.14 l/kg, suggesting that the drug might be protein bound. However, both the elimination half-life and MRT were short at 60 minutes. Clearance was 0.13 ± 0.04 l/kg/hr. The dose necessary to maintain the breakpoint MIC of 8 μg/mL for at least two-thirds of the dosing interval (above 2 μg/mL for the entire interval) (for humans) in dogs was recommended by the author to be 40 mg/kg every 6 hours.28
Ceftazidime is a third-generation drug characterized by an elimination half-life of 0.8 hours in dogs. After subcutaneous injection, Tmax occurs at 1 hour after administration of 30 mg/kg. When given an initial dose of 4.4 mg/kg followed by a constant-rate infusion of 4.1 mg/kg/hr for 36 hours, Cmax at steady state is 22.2 μg/mL. Total body clearance is 0.19 L/kg/hr.69 The MIC90 for clinical isolates (n = 101) of P. aeruginosa was ≤ 4 μg/mL.69 Using 4μg/mL as the basis for a subcutaneous dose of 30 mg/kg, only 3 half-lives can elapse for T = MIC, indicating a 6-hour dosing interval might be appropriate for Pseudomonas spp. Ceftazidime has been studied in cats (n = 5) after intravenous and intramuscular (30 mg/kg) administration.70 After intravenous administration, the Vd was 18 ± 0.04 L/kg; protein binding was not described. Plasma clearance was 0.19 ± 0.08 L/hr/kg, and elimination half-life was 0.77 ± 0.06 hour. After intramuscular administration, bioavailability was 82.47 ± 4.37%, resulting in a Cmax of 89.42 ± 12.15 μg/mL, at a Tmax of approximately 30 minutes. The authors indicated that for an 8- to 12-hour dosing interval, T > MIC would range from 35% to 52% of the dosing interval for intravenous and 48% to 72% for intramuscular administration for isolates with an MIC ≤ 4 μg/mL.
Ceftiofur is a third-generation drug approved for use in dogs for treatment of urinary tract infections. It has been studied at 0.22, 2.2, and 4.4 mg/kg administered subcutaneously in dogs (n = 9).71 PDCs increase proportionately (see Table 7-1). It has a relatively long half-life compared with other cephalosporins, reflecting, in part, its active metabolite. Accordingly, a longer dosing interval is likely to be more reasonable for ceftiofur compared with the first-generation drugs. When administered subcutaneously, peak PDCs (Cmax) were 1.66 ± 0.2, 8.91 ± 6.42, and 27 ± 1 μg/mL at 0.22, 2.2, and 4.4 mg/kg, respectively.71 At the Cmax of approximately 9 μg/mL at a dose of 2.2 mg/kg, targeting T > MIC of 50%, the highest MIC that can be treated at 12-hour intervals is 4 μg/mL. At 4.4 mg/kg administered subcutaneously, the Cmax disproportionately increases to 29 μg/mL, and the highest MIC that could be treated using the same targets is 16 μg/mL, which actually exceeds the MICBP (≥8 μg/mL). Urine concentrations were also reported for ceftiofur bioactivity in the dog. At 24 hours, urine concentrations at 2.2 and 4.4 mg/kg were 8.1 and 29.6 μg/mL, respectively. These concentrations surpassed the the MIC90 for E. coli (4.0 μg/mL) and P. mirabilis (1.0 μg/mL).71
Tissue kinetics of cefpodoxime compared with cephalexin have been described in dogs.59a The free and thus diffusible fraction of drug in plasma ranged from 9% to 34%. Maximum drug concentrations after administration of 8.5 mg/kg (single dose) in dogs (n = 6) was (extrapolated from plot) approximately 10 μg/mL free drug (33±7 μg/mL total) in plasma compared with 4.3 +1.9 in interstitial fluid, suggesting less than 50% of the drug in plasma reaches interstitial tissues. Unbound AUC in plasma was not provided, but the disappearance half-life of cefpodoxime from interstitial fluid was twice as long as that from plasma (10 + 3 hours versus 5.6 + 0.9 hours, respectively). The reason for this difference is not clear, although factors that influence diffusibility from tissue into serum might also influence antibacterial activity potentially precluding drug efficacy. Nonetheless, on the basis of these data, interstitial concentrations of cefpodoxime exceeded the MIC90 of S. intermedius and E. coli as reported on the package insert for 24 hours.59a This is in contrast to cephalexin, which is <20% bound to plasma proteins and for which interstitial concentrations exceeded the MIC90 for S. pseudintermedius (as reported by Stegemann20) for 12 hours but did not achieve the MIC90 for E. coli.
Cefovecin (third-generation) is the newest cephalosporin to be approved in dogs at the time of this publication. Its PD and PK have been very well described including either concentrations or bioactivity in interstitial fluid in dogs or cats in part because its disposition is complicated by extensive binding to plasma proteins.20,72,73 Accordingly, care must be taken when designing dosing regimens to base decisions on unbound, rather than total, drug. Based on protein-binding studies (microdialysis) at cefovecin concentrations ranging from 10 to 300 μg/mL in dog plasma, 96% to 98% is bound at concentrations below 100 μg/mL, with the fraction increasing to 72% at 200 μg/mL and 56% at 300 μg/mL. Avid protein-binding results in a slow release and a long elimination half-life of 136 or 133 hours when given intravenously or subcutaneously, respectively. Protein-binding also affects Tmax, which does not occur until 6 hours (based on total drug), and the apparent Vd (0.12 L/kg), which is higher than total blood volume but considerably lower than extraceullar fluid volume. Cmax of unbound, active drug approximates about 5 μg/mL. Predicted unbound concentrations suggest that T > MIC90 of, S. pseudintermedius (0.25 μg/mL) occurs at approximately day 12 after dosing 8 mg/kg subcutaneously; however, this is reduced to day 8 on the basis of the lowest unbound concentration predicted by the 95% confidence interval of 1 μg/mL, which is the more prudent statistic to follow (see package insert). For organisms with MIC ≥ 2 μg/mL (see Table 7-9) (e.g., S. aureus, not an approved indication), T > MIC of mean (predicted) unbound drug at approximately 1 to 2 days; however, if based on the lowest (95% confidence interval) predicted unbound concentrations, 2 μg/mL would not be reached in plasma. In contrast, the MIC90 of Streptococcus canis (an approved indication) is much lower (< 0.06 μg/mL); thus T > MIC exceeds 14 days even when based on the lowest predicted unbound concentration in plasma. The same is true for Pasteurella, the approved indication in cats; the targeted T > MIC90 is not reached until 12 days after treatment.
Studies of unbound cefovecin in tissue have been published using tissue cage models in dogs.72 The studies demonstrate that unbound cefovecin effectively moves from plasma into tissues, as indicated by antibacterial activity against S. pseudintermedius across time). After 8 mg/kg administered subcutaneously in dogs, cefovecin (total) Cmax (total, μg/mL) was 116, 32, and 40 in plasma, transudate, and exudate, respectively, with elimination half-life from transudate similar to that in plasma (147 hours and 136 hours, respectively). Antibacterial activity was detectable in transudate at 4 hours; however, Tmax of cefovecin antibacterial activity did not occur until approximately 2 days. Interestingly, antibacterial activity in transudate actually exceeded antibacterial activity in plasma at all time points after 8 hours and far exceeded it from day 5 forward. Peak antibacterial effects for S. pseudintermedius persisted in transudate until day 10 after injection, with log 2 reduction in CFUs still present at day 18; activity was gone by day 21.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


