Selection of the appropriate antifungal drug and dosage for treatment of systemic fungal infections can be difficult. Choice of an antifungal drug regimen based on susceptibility data is problematic for several reasons.64 The widely divergent growth properties of yeasts and filamentous fungi make in vitro cultivation difficult. There is no standardized or accurate means to determine quantitative growth of filamentous fungi. Antifungal susceptibility data exist39; however, correlation between susceptibility data and clinical efficacy has not been well studied. Pharmacokinetic determinations of antifungal drug concentrations in various tissues or body fluids of dogs and cats is minimal. Although antifungal drugs appear to be fungicidal in vitro, most all are fungistatic in vivo.64 Therefore, in most instances, host immune responses must be relied on to successfully eradicate the organisms. As a general guideline, Table 55-1 lists various systemic antifungal drugs and the circumstances in which they are used. Chapters 56 to 69 provide information on their use for specific diseases, and details on dosage and usage are given in the Drug Formulary in the Appendix. TABLE 55-1 Systemically Used Antifungal Drugs CNS, Central nervous system; IV, intravenous; KI, potassium iodide; NaI, sodium iodide; PO, by mouth; SC, subcutaneous. For additional information on drugs and dosages, see the Drug Formulary in the Appendix. aMicafungin and anidulafungin have similar recommendations; see text for further information on these drugs. Griseofulvin is deposited in the epidermal layers of the skin and dermal appendages as they are formed. Several weeks of therapy are required for complete drug distribution throughout cell layers and for inhibition of fungal growth. Resistance rarely develops during therapy. Despite its teratogenic effects, griseofulvin does not have deleterious effects on semen quality in dogs.188 A solution of 20% sodium iodide or 100% (supersaturated) potassium iodide has been given orally to treat cutaneous sporotrichosis (see Chapter 61). The mechanism of action of the iodides is uncertain because they are not directly toxic to Sporothrix species in vitro. Iodides are all less toxic than amphotericin B (AMB), which should be reserved as a second-choice drug or for disseminated sporotrichosis. Iodism, manifested clinically as dermal eruption with hair loss, may occur as a result of therapy. AMB, a lipophilic polyene isolated from Streptomyces nodosus, binds to sterols in cell membranes of eukaryotic organisms and causes increased permeability and leakage of nutrients and electrolytes. AMB has greater binding affinity for ergosterol, the major sterol of fungal cell membranes, than for cholesterol, which is present in mammalian host cells. Because it is poorly absorbed across the GI mucosa or skin, AMB must be given parenterally (usually intravenously) for the treatment of systemic mycoses. Solutions or topical formulations have been used for treatment of mucosal or dermal infections. Results from in vivo studies against fungal infections show superior efficacy when the drugs are given every three days as compared to daily dosing, and higher peak plasma levels, as compared to minimum inhibitory concentration, are reached.200 Most of the drug is metabolized locally at tissue sites, and lesser amounts are excreted in the urine. Standard IV preparations contain lyophilized AMB combined with the bile salt deoxycholate and buffer in a colloidal state (ABD). ABD has been diluted and given subcutaneously, which appears to delay its absorption and reduce its nephrotoxicity, allowing larger amounts of the drug to be given.106 Lipid-based formulations are now available with lower toxicity (Table 55-2); however, merely giving ABD in a fat emulsion did not alter nephrotoxicity.147,190 In contrast, commercially prepared lipid-encapsulated formulations are taken up well into organs of the mononuclear phagocyte system where the organism resides; they accumulate at relatively low levels in the kidneys.9,93 Lipid-based formulations can be given at a higher dose and may be effective in treating resistant or ABD-unresponsive mycoses.132 The overall dose rate for these drugs can be higher because of their lower toxicity. Lipid formulations of AMB are not as potent as conventional AMB on a mg/kg basis.3 However, their distribution may differ, because brain tissue concentrations of liposomal AMB (L-AMB) are 6- to 10-fold higher than those of other AMB preparations56 and are an important determinant of efficacy in central nervous system (CNS) infections.86 The cost of these lipid formulations may be prohibitive for certain veterinary patients. Occasionally, the AMB formulations are used for topical therapy of mycotic disease. (See the Drug Formulary in the Appendix for information about the various preparations and specific information regarding AMB use.) TABLE 55-2 Comparison of Clinical Formulations of Amphotericin Ba ABCD, Amphotericin B with cholesteryl sulfate; ABD, colloidal dispersion of amphotericin B and the bile salt, deoxycholate; ABLC, amphotericin B with lipid complex; AMB, amphotericin B; H, hemisuccinate vesicles; L-AMB, amphotericin B with encapsulated unilamellar liposomes; LD50, lethal dose killing 50% of treated mice; LIP, liposomal complex; ND, no data; PEG, polyethylene glycol. aFor additional information, see the Drug Formulary in the Appendix. bApothecon, Division of Bristol Myers-Squibb, Princeton, NJ. cAstellas Pharma US, Deerfield IL, in cooperation with Gilead Sciences Inc. dThree Rivers Pharmaceuticals, Warrendale, PA. eEnzon Pharmaceuticals, Bridgewater, NJ. fInvestigational only; not commercially available. The fungal organisms affected by AMB are listed in Table 55-1. Zygomycetes (family Mucoraceae) are variably susceptible, and Aspergillus spp., Fusarium spp., Trichosporon beigelii, Pseudallescheria boydii and some species of Candida are usually resistant.64 Acquired resistance has also developed by Candida, causing infections in some humans during the course of treatment. Because higher effective AMB dosage levels can be achieved, lipid-based formulations are more effective against zygomycosis and fusariosis, and they might be the first choice for treating these diseases.69 Hamycin is another polyene that is effective against Candida, Cryptococcus, Histoplasma, Blastomyces, and Aspergillus species. It has been given orally, topically, and intraperitoneally to treat various mycotic conditions, although toxicity does occur. A dog with disseminated aspergillosis was treated with some success.83 Liposomal encapsulation of this drug has been associated with increased efficacy and reduced toxicity.120 Nystatin is another polyene and is applied topically; however, parenteral formulations are under development (see Topical Antifungals, later). Flucytosine, or 5-fluorocytosine (FCY), is a fluorinated pyrimidine originally synthesized as an antineoplastic agent. It is converted into 5-fluorouracil within target cells. It interferes with pyrimidine metabolism via the enzyme thymidylate synthase and resultant DNA synthesis in yeasts. Most filamentous fungi and mammalian cells lack the associated enzymes for its intracellular incorporation. In contrast to studies with AMB, efficacy of FCY is increased when small dose levels are given with greater frequency.4 With lower serum levels, toxicity might be reduced; however, penetration of difficult-to-reach areas such as the CNS might be compromised. FCY is effective against Cryptococcus species, Candida species, and other yeasts but has little or no effect on other deep mycotic agents or on Aspergillus species. Localized candidiasis or cryptococcal infections respond best, but resistance to FCY frequently develops during therapy. For this reason, the drug is always given in combination with AMB. Skin eruptions similar to those caused by azoles have been observed in treated dogs.108 GI and myelosuppressive effects are the primary side effects observed in people (see the Drug Formulary in the Appendix). Synthetic azole derivatives were originally produced as broad-spectrum anthelmintics with activity against some gram-positive bacteria and protozoa. Like AMB, they inhibit sterol synthesis via fungal cell cytochrome P450–dependent enzyme, lanosterol 14α-demethylase, as one of their main effects, but they are generally less toxic. The enzyme is present in most species of molds and yeasts and in the hemoprotozoan Leishmania species. It is lacking in Pythium spp. Azoles also have relatively low levels of inhibition of mammalian sterol synthesis. Hence, side effects of reduced testosterone, cortisol, androgen, or cholesterol may be observed with some of the compounds. They also inhibit nucleic acid, triglyceride, and fatty acid synthesis and alter oxidative enzyme biochemistry. At low concentrations they are fungistatic, and at higher concentrations, which cannot be achieved systemically, they are fungicidal. When given systemically, they vary in pharmacokinetic parameters such as oral bioavailability, protein binding, plasma clearance, and volume of distribution (see the Drug Formulary in the Appendix). Efficacy of the azoles seems dependent on the total amount of drug given in a 24-hour period, rather than the interval of administration.2 All azoles undergo hepatic metabolism by cytochrome P450 enzymes, and this means of elimination can lead to drug interactions. Azole levels are decreased by coadministered drugs that reduce azole absorption or accelerate their metabolism (Table 55-3). Unexpected toxicity of coadministered drugs may result because of azoles delaying their metabolism (see Table 55-3). TABLE 55-3 Drug Interactions of Commonly Used Antifungal Drugs37,54,54 FCZ, Fluconazole; ITZ, itraconazole; KTZ, ketoconazole; VCZ, voriconazole. aAny antacid that raises gastric pH will interfere with absorption of azoles. bThis interaction is used to increase the drug concentration of ciclosporin to help reduce the required dosages for treating perianal fistulas in dogs. Of the imidazole group, thiabendazole was initially recognized as being effective in treating human dermatophytoses. Imidazoles may have enhanced topical efficacy because they may cause direct fungal membrane lysis in addition to their effect on ergosterol synthesis.64 Miconazole, clotrimazole, and enilconazole are available in creams and lotions and can be formulated into solutions for topical treatment of fungal infections of the skin, such as dermatophytosis and candidiasis (see Topical Antifungals in this chapter and the Drug Formulary in the Appendix). However, oral therapy with an azole, griseofulvin, or terbinafine (see later discussion) is needed for nail infections. Topical application of imidazoles, including enilconazole or clotrimazole, has been used in the treatment of nasal and sinus infections with aspergillosis. Of the orally administered azoles, KTZ is an imidazole, whereas newer drugs (ITZ and FCZ) are triazoles. The orally administered azoles are becoming widely used in human and veterinary medicine to treat systemic and opportunistic fungal infections. Oral administration has been preferred in many situations because it is easier than intravenous administration of AMB. In vitro susceptibility testing of the drugs shows variable efficacy against different fungi, but it does not always parallel in vivo efficacy. Pretreatment or combination therapy using azoles with AMB has not been recommended because, in experimentally infected rodents, reduced treatment efficacy, presumably from a reduction in ergosterol concentrations, has been reported.104 Further evaluation of this potential inhibition is needed. Azoles have also been used in human medicine as prophylactic treatment for fungal superinfections in immunocompromised patients. However, breakthrough infections with azole-resistant fungi in the class Mucorales, causing zygomycosis, have been a concern with routine prophylaxis.134 GI problems are the most common side effects observed with KTZ in dogs.115 Occasional nausea, partial anorexia, and vomiting in dogs can be overcome if the drug is given with meals and by dividing the daily dose into three or four administrations. Additional side effects of KTZ are similar to those of other azoles. Frequently, reversible subclinical increases in activities of hepatic aminotransferases and alkaline phosphatase in blood occur as manifestations of hepatotoxicity. Less commonly, clinical hepatitis develops, and if treatment continues or dosages are high enough, it may be fatal. Histologic findings in affected animals include enlarged portal tracts, bile duct proliferation, and infiltration with mononuclear cells. KTZ may produce endocrine dysfunction by suppressing testosterone and cortisol synthesis. KTZ is most effective in vitro against yeast and dimorphic fungi such as Candida, Malassezia, Coccidioides, Histoplasma, and Blastomyces species and is less effective against Cryptococcus, Sporothrix, and Aspergillus species. Evidence suggests that in rapidly progressing systemic mycoses, such as in many cases of blastomycosis, patients should first be treated with AMB and then maintained on KTZ (see Amphotericin B and Other Polyenes in this chapter). The action of KTZ alone often occurs so slowly (over 5 to 10 days) that the disease progresses before the drug has a chance to take effect. In contrast, KTZ has been given alone in dogs and cats to successfully treat coccidioidomycosis and some cases of histoplasmosis and cryptococcosis, although relapses are common when host defenses are inadequate. Preference for more active and generally less toxic ITZ or FCZ has been apparent for treatment of these infections. If underlying immunosuppression is thought to be responsible for the fungal disease, concurrent treatment with AMB is recommended. No evidence suggests that KTZ provides synergistic effects with other antifungals except AMB, and even that evidence is inconclusive. (For dosing and detailed information on administration, see the Drug Formulary in the Appendix, and Chapters 56 to 68.) ITZ is a lipophilic, highly protein bound, broad-spectrum azole with more potent in vitro activity than KTZ against Candida species, Aspergillus species, and dermatophytes. Its bioavailability is variable, being affected by a variety of influences; therefore, therapeutic monitoring has been used for this drug.27a Being highly protein bound, it does not enter the CNS, nor is it eliminated in the urine. It can be given orally or parenterally and is widely distributed in body tissues other than the CNS and aqueous body fluids. Concentrations in parenchymal organs, musculoskeletal tissues, skin, and nails are greater than in plasma. For reasons of better spectrum and lower side effects, as compared to those of KTZ, it is often preferred for non-CNS infections with Blastomyces, Histoplasma, Coccidioides, and Aspergillus. The best success in treating human patients (in decreasing order) has been in those with paracoccidioidomycosis, blastomycosis, sporotrichosis, noninvasive aspergillosis, meningeal cryptococcosis, and aspergilloma. Some patients with zygomycosis or other fungal infections also respond to ITZ treatment. In small animal practice, it has been most effective in treating blastomycosis (see Chapter 57), histoplasmosis (see Chapter 58), cryptococcosis (see Chapter 59), and coccidioidomycosis (see Chapter 60). ITZ seems to be less toxic than KTZ, probably because ITZ more selectively inhibits fungal rather than mammalian enzymes. Therefore, unlike KTZ, ITZ has minimal effects on androgen or cortisol metabolism. Higher dosages should be expected to cause side effects similar to those of KTZ. Dosage adjustments do not appear to be necessary in the presence of renal dysfunction. Bioavailability after oral administration of capsules is erratic, but it can be maximized by giving ITZ with food or fats. An oral solution improves absorption, especially in cats.14 An intravenous formulation that is being discontinued in many countries was used for treatment of severe resistant infections such as aspergillosis. High steady-state plasma concentrations could be reached without the loading period required for oral formulations. ITZ is metabolized by the liver into primarily active metabolites; however, ITZ causes interactions with cytochrome P450–metabolized drugs (see the Drug Formulary in the Appendix). FCZ (UK-49,858) is an orally active agent that has been used to treat systemic mycoses, including cryptococcal meningitis, blastomycosis, and histoplasmosis, and to treat superficial infections, including candidiasis and dermatophytosis. The advantages of FCZ are that it is well absorbed orally and has a relatively long elimination half-life (12 hours in cats, 14 hours in dogs), making it valuable for treating susceptible fungal infections in various tissues. Its high water solubility and low protein binding facilitate its wide distribution and penetration of most tissues and body fluids. FCZ crosses the blood-brain and blood–cerebrospinal fluid barriers better than the older azole derivatives. It is effective for treating cryptococcosis because of its CNS penetration.133 Voriconazole (VCZ), an FCZ derivative described in the following section, also has excellent CNS penetration. Based on in vitro susceptibility testing, FCZ is generally less active than ITZ, especially against filamentous fungi. However, FCZ is more effective in vivo, presumably because of its extensive distribution throughout the body. It seems to be less toxic than KTZ. Against dimorphic fungi, it is most active in decreasing order against Coccidioides immitis, Histoplasma capsulatum, and Blastomyces dermatitidis. It is relatively active against Cryptococcus spp. and has frequently been used for treatment of infections with this yeast. It is not very effective against filamentous fungi such as Aspergillus species. Most (70%) FCZ is excreted unchanged in the urine, which makes it valuable for treating lower urinary tract yeast infections caused by Candida species. With renal parenchymal involvement, other antifungals may be as effective as FCZ or even more effective, because tissue concentrations in the extracellular space of the kidney, which are low for FCZ, are more important than urine concentrations.3 First choice recommendations for FCZ use would be to treat infections caused by Candida or Cryptococcus with CNS involvement. VCZ (Pfizer Ltd, Sandwich, UK) is a synthetic derivative of FCZ. VCZ is licensed for treatment of drug-resistant Candida and Aspergillus infections in people. It is the first among a second generation of triazoles, including posaconazole (PSZ), to be licensed. These three drugs have enhanced broad-spectrum antifungal activity against Candida, Trichosporon, Cryptococcus, Histoplasma, Blastomyces, Aspergillus, and Fusarium species and against some zygomycetes. The drugs have also demonstrated potent therapeutic efficacy in treatment of invasive pulmonary aspergillosis and disseminated and oropharyngeal candidiasis in experimental animals. Candida spp. from dogs and cats were more susceptible to VCZ than ITZ.129 VCZ can penetrate the CNS barrier with higher levels in brain tissue versus plasma. VCZ has been more effective than AMB in the initial treatment of invasive aspergillosis in humans,68,137 but it has shown some tendency to produce hepatotoxicity in humans.146 Fungi are more likely to develop resistance to VCZ when the initial drug susceptibility was marginal. Recommendations would be to consider this drug for severe or resistant Candida or Aspergillus infections or those due to azole-resistant genera such as Scedosporium and Fusarium. Neurotoxicity or hepatotoxicity can develop with use of this drug.50a,146a (See the Drug Formulary in the Appendix for additional information on VCZ.) PCZ (previously SCH 56592; Schering-Plough, Kenilworth, NJ), a hydroxylated analogue of ITZ, is least active against Candida species that are resistant to FCZ or ITZ. However, its in vitro activity against Aspergillus species is greater than that of ITZ and VCZ. Bioavailability after oral administration is somewhat variable.105a It is widely distributed to tissue and fluids including pulmonary alveolar cells, but does not penetrate the CNS. PCZ has been formulated as an oral tablet and suspension and has been licensed for use in human medicine. Preliminary trials in rodents with experimental pulmonary invasive aspergillosis have shown PCZ to be superior to AMB or other triazoles in reducing fungal burden. Its primary use would be a second line of treatment for resistant infections with filamentous or yeast fungi. For further information on use in dogs and cats, see the Drug Formulary in the Appendix. Allylamines are synthetic fungicidal agents that reversibly inhibit squalene cyclase, an enzyme in ergosterol synthesis. Butenafine and naftifine are topically used members of this class of antifungal drugs. Terbinafine is an orally administered drug that has good in vitro activity against Aspergillus spp., Fusarium spp., and other filamentous fungi but variable activity against yeasts. It has been extremely effective for treating people with chronic dermatophytosis of the skin or nails, sporotrichosis, and superficial yeast infections. It has been used to treat feline dermatophytosis (Chapter 56) and sporotrichosis (Chapter 61). Compared to griseofulvin, it is more effective, less toxic, and requires shorter treatment intervals. High concentrations are reached in dermal tissues. Improvement or resolution of onychomycosis usually occurs after 3 to 6 months of therapy. Side effects have been minimal. This drug has potential value for treating canine and feline onychomycosis. Terbinafine has been used alone or in combination with azoles and AMB for treatment of aspergillosis and other filamentous fungal infections; however, its activity alone against systemic infections is less impressive (see the Drug Formulary in the Appendix).
Antifungal Chemotherapy
Systemic Antifungals
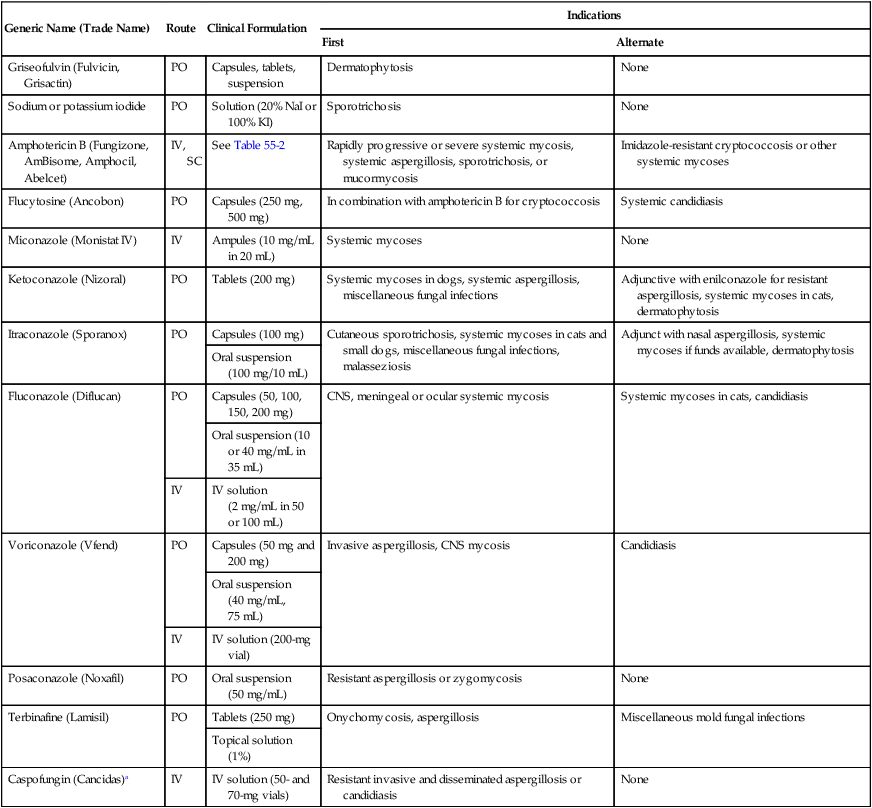
Generic Name (Trade Name)
Route
Clinical Formulation
Indications
First
Alternate
Griseofulvin (Fulvicin, Grisactin)
PO
Capsules, tablets, suspension
Dermatophytosis
None
Sodium or potassium iodide
PO
Solution (20% NaI or 100% KI)
Sporotrichosis
None
Amphotericin B (Fungizone, AmBisome, Amphocil, Abelcet)
IV, SC
See Table 55-2
Rapidly progressive or severe systemic mycosis, systemic aspergillosis, sporotrichosis, or mucormycosis
Imidazole-resistant cryptococcosis or other systemic mycoses
Flucytosine (Ancobon)
PO
Capsules (250 mg, 500 mg)
In combination with amphotericin B for cryptococcosis
Systemic candidiasis
Miconazole (Monistat IV)
IV
Ampules (10 mg/mL in 20 mL)
Systemic mycoses
None
Ketoconazole (Nizoral)
PO
Tablets (200 mg)
Systemic mycoses in dogs, systemic aspergillosis, miscellaneous fungal infections
Adjunctive with enilconazole for resistant aspergillosis, systemic mycoses in cats, dermatophytosis
Itraconazole (Sporanox)
PO
Capsules (100 mg)
Cutaneous sporotrichosis, systemic mycoses in cats and small dogs, miscellaneous fungal infections, malasseziosis
Adjunct with nasal aspergillosis, systemic mycoses if funds available, dermatophytosis
Oral suspension (100 mg/10 mL)
Fluconazole (Diflucan)
PO
Capsules (50, 100, 150, 200 mg)
CNS, meningeal or ocular systemic mycosis
Systemic mycoses in cats, candidiasis
Oral suspension (10 or 40 mg/mL in 35 mL)
IV
IV solution (2 mg/mL in 50 or 100 mL)
Voriconazole (Vfend)
PO
Capsules (50 mg and 200 mg)
Invasive aspergillosis, CNS mycosis
Candidiasis
Oral suspension (40 mg/mL, 75 mL)
IV
IV solution (200-mg vial)
Posaconazole (Noxafil)
PO
Oral suspension (50 mg/mL)
Resistant aspergillosis or zygomycosis
None
Terbinafine (Lamisil)
PO
Tablets (250 mg)
Onychomycosis, aspergillosis
Miscellaneous mold fungal infections
Topical solution (1%)
Caspofungin (Cancidas)a
IV
IV solution (50- and 70-mg vials)
Resistant invasive and disseminated aspergillosis or candidiasis
None
Griseofulvin
Iodides
Amphotericin B and Other Polyenes
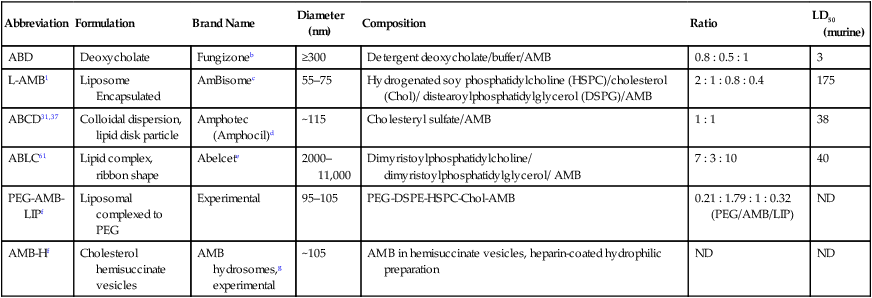
Abbreviation
Formulation
Brand Name
Diameter (nm)
Composition
Ratio
LD50 (murine)
ABD
Deoxycholate
Fungizoneb
≥300
Detergent deoxycholate/buffer/AMB
0.8 : 0.5 : 1
3
L-AMB1
Liposome Encapsulated
AmBisomec
55–75
Hydrogenated soy phosphatidylcholine (HSPC)/cholesterol (Chol)/ distearoylphosphatidylglycerol (DSPG)/AMB
2 : 1 : 0.8 : 0.4
175
ABCD31,37
Colloidal dispersion, lipid disk particle
Amphotec (Amphocil)d
~115
Cholesteryl sulfate/AMB
1 : 1
38
ABLC61
Lipid complex, ribbon shape
Abelcete
2000–11,000
Dimyristoylphosphatidylcholine/ dimyristoylphosphatidylglycerol/ AMB
7 : 3 : 10
40
PEG-AMB-LIPf
Liposomal complexed to PEG
Experimental
95–105
PEG-DSPE-HSPC-Chol-AMB
0.21 : 1.79 : 1 : 0.32 (PEG/AMB/LIP)
ND
AMB-Hf
Cholesterol hemisuccinate vesicles
AMB hydrosomes,g experimental
~105
AMB in hemisuccinate vesicles, heparin-coated hydrophilic preparation
ND
ND
Flucytosine
Azole Derivatives
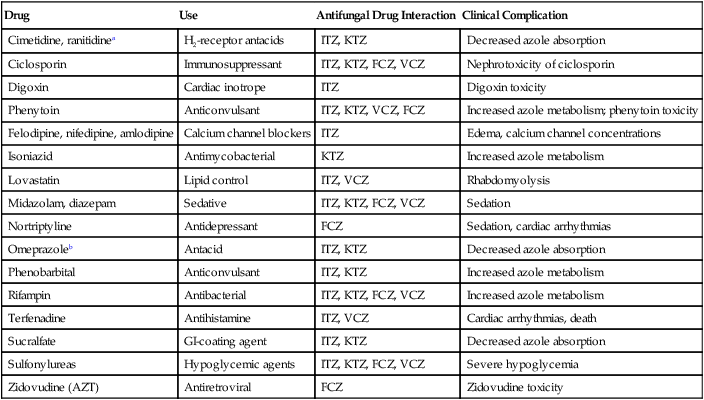
Drug
Use
Antifungal Drug Interaction
Clinical Complication
Cimetidine, ranitidinea
H2-receptor antacids
ITZ, KTZ
Decreased azole absorption
Ciclosporin
Immunosuppressant
ITZ, KTZ, FCZ, VCZ
Nephrotoxicity of ciclosporin
Digoxin
Cardiac inotrope
ITZ
Digoxin toxicity
Phenytoin
Anticonvulsant
ITZ, KTZ, VCZ, FCZ
Increased azole metabolism; phenytoin toxicity
Felodipine, nifedipine, amlodipine
Calcium channel blockers
ITZ
Edema, calcium channel concentrations
Isoniazid
Antimycobacterial
KTZ
Increased azole metabolism
Lovastatin
Lipid control
ITZ, VCZ
Rhabdomyolysis
Midazolam, diazepam
Sedative
ITZ, KTZ, FCZ, VCZ
Sedation
Nortriptyline
Antidepressant
FCZ
Sedation, cardiac arrhythmias
Omeprazoleb
Antacid
ITZ, KTZ
Decreased azole absorption
Phenobarbital
Anticonvulsant
ITZ, KTZ
Increased azole metabolism
Rifampin
Antibacterial
ITZ, KTZ, FCZ, VCZ
Increased azole metabolism
Terfenadine
Antihistamine
ITZ, VCZ
Cardiac arrhythmias, death
Sucralfate
GI-coating agent
ITZ, KTZ
Decreased azole absorption
Sulfonylureas
Hypoglycemic agents
ITZ, KTZ, FCZ, VCZ
Severe hypoglycemia
Zidovudine (AZT)
Antiretroviral
FCZ
Zidovudine toxicity
Ketoconazole
Itraconazole
Fluconazole
Voriconazole
Posaconazole
Terbinafine
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Veterian Key
Fastest Veterinary Medicine Insight Engine
