Inflammatory cell infiltrates in the orbital tissue of CD3+ T cells and macrophages
Pathogenic autoantibodies to the thyrotropin receptor
Chemosis characterized by histopathological signs of inflammatory, congested blood vessels, and accumulation of mast cells
Association in family members favors contribution of genetic factors
Success of immunotherapies targeting immune cells, e.g., CD20 on B cells by rituximab
To define mechanisms underlying GO requires experimental models that manifest the clinical course and pathology of the disease. However, no spontaneous models of Graves’ disease and the associated orbital manifestations similar to GO are available, as these conditions appear to occur spontaneously only in humans (Aliesky et al. 2013) . Even nonhuman primates, species that are mostly similar and evolutionary closest to humans, do not appear to succumb to spontaneous autoimmune hyperthyroidism (McLachlan et al. 2011) . Since in humans, GO occurs commonly in patients with ongoing autoimmune Graves’ hyperthyroid disease, any development of a model for the orbital condition has to have a corollary of a model for experimental Graves’ disease.
Mouse models of induced Graves’ disease have been developed over the past two decades, and although some reported orbital manifestations, none have been verified in independent laboratories and have proved to be disappointing failures (Baker et al. 2005; Dağdelen et al. 2009; Wiesweg et al. 2013) . More recently, the adoptive transfer of TSHR primed spleen cells from TSHR knockout mice (which fail to develop tolerance to the TSHR) to athymic nu-/nu- mice showed small histopathological lesions in the orbital tissue of some animals, but clinically overt disease was not present (Nakahara et al. 2012) .
In this chapter, we discuss the development and latest research on a mouse model of GO that recapitulates a number of features to those present in human disease. However, it becomes necessary to briefly describe mouse models of Graves’ disease to comprehend the development of the model for GO. For more detail on models of Graves’ disease, the reader is referred to recent reviews that describe the multiple models, which recapitulate different aspects of the disease (Dağdelen et al. 2009; Wiesweg et al. 2013) .
8.2 Experimental Models of Graves’ Disease
Generally, experimentally induced models of autoimmune disease are induced by immunization with the potential self-antigen implicated in the autoimmune condition together with an adjuvant (termed active induction) in genetically susceptible animals (Rose 1988). Such methods allow studies on both genetic and molecular investigations, as well as T cell regulation, and have added much to the understanding of autoimmune conditions (Kong 1997) . However, initial attempts to establish experimental Graves’ disease by immunization with characterized preparations of purified human TSHR (hTSHR) protein in an adjuvant did not lend to the induction of thyroid stimulating antibodies and hyperthyroid disease, although large amounts of antibody and T cell reactivity to the receptor were produced (Carayanniotis et al. 1995; Wang et al. 1998; Banga 2007) .
In vivo delivery of TSHR was the key to developing successful mouse models of Graves’ disease. Several induction models for Graves’ disease in inbred mice have been described, with different disease incidence, course, and pathological features (Dağdelen et al. 2009; Wiesweg et al. 2013) .
Almost all mouse models of Graves’ disease have relied on the BALB/c (H-2d) inbred mouse strain with the highest degree of disease susceptibility (Dağdelen et al. 2009; Wiesweg et al. 2013) . To establish efficient in vivo expression of TSHR, genetic delivery by plasmids encoding the extracellular region of hTSHR, termed hTSHR A-subunit, was evaluated. However, these early plasmid DNA delivery models proved to be unreliable in terms of low disease incidence and reproducibility (Pichurin et al. 2001; Rao et al. 2003; Banga 2007) . The most commonly used model, replicated in a number of laboratories, relied on adenovirus-mediated delivery of hTSHR A-subunit cDNA (Chen et al. 2003) , originally based on the innovative findings of Nagayama and colleagues using a full-length hTSHR coding plasmid for immunization (Nagayama et al. 2002). The earlier mouse models of Graves’ disease, including the adenovirus model, did not lead to any visible signs of overt GO (Nagayama et al. 2002; Gilbert et al. 2006) . This raised questions whether other autoantigens, such as the insulin growth factor receptor 1 (IGF-1R), by itself or in association with TSHR was the reason for the lack of orbital pathology (Wiersinga 2012) . A notable feature of Graves’ disease models is short-lived immune responses, as the anti-TSHR response declines soon after the cessation of immunization (Gilbert et al. 2006; McLachlan et al. 2012) . It is likely that in these models the short duration of anti-TSHR responses results in acute phase of the disease, which is sufficient to induce hyperthyroid disease, but inadequate for the development of pathological events leading to remodeling of orbital tissue. Thus, development of protocols to induce a long and sustained immune response to TSHR in animals undergoing experimental Graves’ disease leading to chronic hyperthyroidism was our rationale for attempts to develop orbital manifestations in the model.
8.3 Experimental Model of Graves’ Orbitopathy
Initial Attempts to Induce Chronic Hyperthyroidism in Mice
Earlier studies attempted to produce chronic stimulation of the thyroid gland by growing hybridoma cells secreting thyroid stimulating antibody in vivo in the peritoneal cavity of mice, but have the drawback that it is difficult to know the amount of monoclonal antibodies (mAb) available in vivo, nor the degree of TSHR stimulation resulting in hyperthyroidism (Ando et al. 2004) . Our studies of atypical chronic hyperthyroid disease relied on regular weekly intravenous transfer over several weeks, of microgram quantities of mAb with strong thyroid stimulating activity (Gilbert at al. 2006; Flynn et al. 2007) . These studies in humanized HLA-DRB1*0301 transgenic mice in the NOD genetic background resulted in achieving the casual relationship between chronic stimulation and resultant hyperthyroidism and goiter, but no visible signs of orbital manifestations were apparent (Flynn et al. 2007) .
Genetic delivery of hTSHR plasmid by in vivo electroporation reported the longevity of the induced immune response to hTSHR (Kaneda et al. 2007) . We refined this protocol using an hTSHR A-subunit plasmid with caliper electrodes for close-field electroporation by delivery of brief, intense electrical pulses to shaved quadricep leg muscle and surrounding tissue which leads to temporary opening of the cell membrane to allow efficient plasmid delivery to inside the cell (Widera et al. 2000) . This approach resulted in a sustained response to TSHR, together with high incidence of induced hyperthyroidism in female BALB/c mice (Zhao et al. 2011). All immune animals developed a strong thyroid stimulating antibody response to hTSHR, accompanied with a large proportion (75 %) of animals with hyperthyroidism. Interestingly, antibodies induced to hTSHR in other murine models of experimental Graves’ disease cross-react weakly with the autologous mouse TSHR, but the special subset of induced thyroid stimulating antibodies cross-react strongly with the stimulating “trigger” determinants on the mouse TSHR to activate the receptor in vivo leading to hyperthyroidism (Nakahara et al. 2010) . Histopathological examination of the orbital tissue gave the first indications of orbital inflammation and fibrosis in the model (Zhao et al. 2011). In principle, our model replicates the situation in Graves’ disease patients by an ongoing autoimmune response and continuous chronic stimulation of thyroid gland .
IGF-1R has been implicated as a second antigen in GO. Autoantibodies to IGF-1R have been identified in patients with GO (Weightman et al. 1993), although other recent intensive searches for these antibodies have failed to support these findings (Wiersinga 2012; Minich et al. 2013; Varewijck et al. 2013) . Our studies using an IGF-1R α-subunit coding plasmid for immunization by close-field electroporation in BALB/c mice failed to induce any pathology in immune animals, although robust anti-IGF-1R antibodies were induced (Zhao et al. 2011; Moshkelgosha et al. 2013) . Thus, the role of IGF-1R in GO remains challenging. Interestingly, recent studies provide evidence for receptor cross-talk between G-protein coupled receptors and tyrosine kinase receptors (Gavi et al. 2006) . Indeed, such cross-talk has recently been reported between TSHR and IGF-1R in orbital fibroblasts that may provide an explanation for the discrepant findings on anti-IGF-1R antibodies in GO (Krieger et al. 2015) .
Development of a Bona-Fide Model of Graves’ Orbitopathy
In a further development of the electroporation model of experimental Graves’ disease (Zhao et al. 2011), we evaluated small changes in the delivery of the plasmid inoculum into the leg muscle and whether these changes correlated with orbital lesions. We also improved the histopathology of the orbital region of the mouse (Smith 2002), by using the optic nerve as an anatomical reference marker in the section (Fig. 8.1, Panel a) (Johnson et al. 2013) . The modifications to the model presented with the development of GO in mice undergoing autoimmunity to hTSHR (Moshkelgosha et al. 2013) .
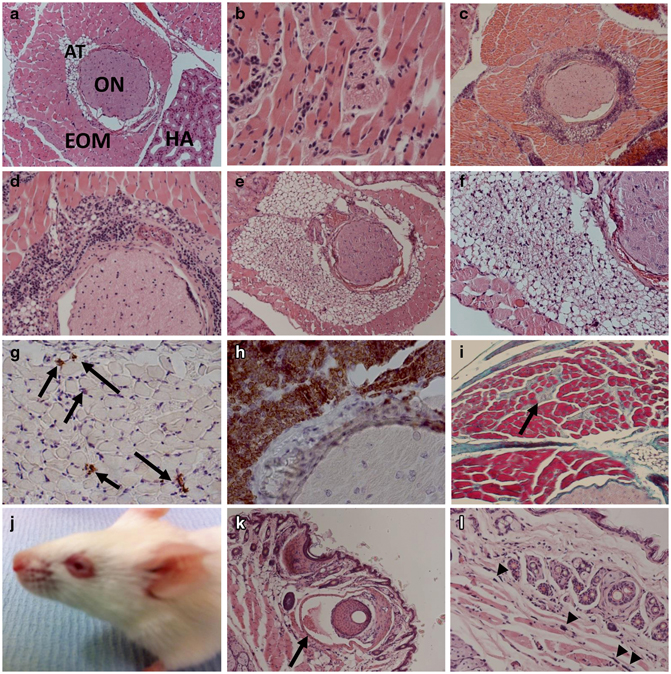
Fig. 8.1
Representative H&E stained sections of orbital tissue from mice undergoing Graves’ ophthalmopathy, following immunization with the hTSHR A-subunit plasmid by close-field electroporation. a Orbital tissue from control b-Gal plasmid immunized mouse (× 100). EOM extraocular muscle; ON optic nerve; AT adipose tissue; HA harderian gland. b Inflammatory infiltrate into orbital muscle tissue (× 100). c Perineural infiltrate around optic nerve (× 100). d Higher magnification (× 200). e Expansion of orbital fat tissue (× 100). f Higher magnification (× 200). g immunohistochemistry (IHC) to show CD3+T cells (arrowed) into orbital muscle tissue (× 200). h IHC showing CD3 + T cells in the perineural infiltrate around optic nerve (× 100). i Fibrosis in retrobulbar tissue at end stage of disease, Masson’s Trichrome-stained section (× 100). j Mouse head to show animal undergoing chemosis. k Chemosis eyelid to show congested blood vessels, arrowed (× 100). l Higher magnification to show accumulation of mast cells, arrow heads (× 200)
The pathology of GO in the model is primarily driven by expansion of the orbital tissue by inflammation and adipogenesis. The majority of the animals show an inflammatory infiltrate into the extraocular muscle (Fig. 8.1, Panel b). In addition, orbital pathology in a few animals was characterized by inflammatory T cells around the optic nerve, but without infiltrate in the optic nerve or in the surrounding retrobulbar tissue (Fig. 8.1, Panels c and d). There were also differences, as some animals exhibit expansion of the retrobulbar adipose tissue, which is sufficiently large enough to separate the orbital muscle fiber bundles (Fig. 8.1, Panels e and f). Immunohistochemical staining identified CD3+ T cells (Fig. 8.1, Panels g and h). There was a distinct absence of B cells in the orbital infiltrate. End-stage disease was characterized by fibrosis (Fig. 8.1, Panel i). Most striking, however, was the finding that some animals also show extraorbital signs of inflammation, such as chemosis (Fig. 8.1, Panel j) with congested blood vessels and accumulation of mast cells (Fig. 8.1, Panels k and l). Further examination of soft tissue in the orbital region by small animal MRI in mice undergoing experimental GO shows clear hypertrophy of the orbital muscles and proptosis of the eye (not shown) (Moshkelgosha et al. 2013) . Thus, the pathology of GO model closely mimics the disease heterogeneity reminiscent of the remodeling of orbital tissue in the human condition (Table 8.2) .
Table 8.2
Clinical outcome in Graves’ orbitopathy model—similarities and differences from the human condition
Similarities |
Inflammatory cell infiltrates in the orbital tissue of CD3+ CD4+ T cells, macrophages, and mast cells (80 % diseased animals) |
Adipogenesis, expansion of orbital adipose tissue (10 % diseased animals) |
Fibrosis of orbital muscle tissue |
Presence of thyrotropin receptor antibodies, with activating or blocking antibodies |
Hyperthyroid or hypothyroid status in animals undergoing Graves’ orbitopathy |
Presence of antibodies to IGF-1R |
Eyelid inflammation, chemosis |
Orbital muscle hypertrophy leading to proptosis |
Differences |
Inflammatory T cells around the optic nerve (5 % diseased animals), but without infiltrate in the optic nerve |
Absence of thyroid inflammation |
Unilateral disease not found, both eyes show the same pathology |
The orbital remodeling in animals undergoing experimental GO was associated with sustained antibody responses to TSHR during the entire course of the disease. The antibodies induced to the TSHR comprise of both thyroid stimulating and thyroid stimulating blocking antibodies and depending on the delivery of the hTSHR plasmid inoculum, results predominantly with immune animals presenting with hyperthyroid disease (Zhao et al. 2011) or both hyperthyroid or hypothyroid disease (Moshkelgosha et al. 2013) . Importantly, our studies emphasize that experimental GO develops under hyperthyroid or hypothyroid conditions, very similar to the situation in human patients (Table 8.2).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


