(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
Readers are bound to wonder why in a book on diabetes we have not discussed blood sugar levels at sufficient length so far and why it is delayed till the 12th chapter. The common perception of diabetes begins with blood sugar and often ends with blood sugar. We saw in Chap. 3 that there are a series of paradoxes associated with the existing theory of glucose homeostasis and the conventionally perceived etiology of hyperglycemia. The alternative picture needs to resolve all the paradoxes. We can attempt to do so at this stage since by now I think sufficient background about the new interpretation has been built on which we can construct a new model of normal glucose regulation and altered glucose dynamics in T2D. Discussing sugar so late in this book also serves as a gesture intended to emphasize that sugar levels in blood are actually not as important in diabetes as generally believed. Raised blood sugar to diabetes is just what fever is to typhoid. It is only one of the many symptoms. However, it is one that is easiest to monitor and therefore the first indication in diagnosis and one that can also be conveniently used to follow the progression of the illness as well as any recovery from it. But that is its limitation. Just as fever is not everything in typhoid, blood sugar is not everything in T2D. Typhoid is an infection, and fever is only a symptom. If typhoid is cured, fever will certainly vanish, and the body temperature will return to normal. The reverse is not true. Fever can be reduced by antipyretic drugs or other physical means. Keeping the body on ice can also reduce the body temperature temporarily. But fever gone is not typhoid gone. The relation of blood sugar to T2D is very similar. Bringing blood sugar back to normal is not an indication of diabetes gone.
Readers are bound to wonder why in a book on diabetes we have not discussed blood sugar levels at sufficient length so far and why it is delayed till the 12th chapter. The common perception of diabetes begins with blood sugar and often ends with blood sugar. We saw in Chap. 3 that there are a series of paradoxes associated with the existing theory of glucose homeostasis and the conventionally perceived etiology of hyperglycemia. The alternative picture needs to resolve all the paradoxes. We can attempt to do so at this stage since by now I think sufficient background about the new interpretation has been built on which we can construct a new model of normal glucose regulation and altered glucose dynamics in T2D. Discussing sugar so late in this book also serves as a gesture intended to emphasize that sugar levels in blood are actually not as important in diabetes as generally believed. Raised blood sugar to diabetes is just what fever is to typhoid. It is only one of the many symptoms. However, it is one that is easiest to monitor and therefore the first indication in diagnosis and one that can also be conveniently used to follow the progression of the illness as well as any recovery from it. But that is its limitation. Just as fever is not everything in typhoid, blood sugar is not everything in T2D. Typhoid is an infection, and fever is only a symptom. If typhoid is cured, fever will certainly vanish, and the body temperature will return to normal. The reverse is not true. Fever can be reduced by antipyretic drugs or other physical means. Keeping the body on ice can also reduce the body temperature temporarily. But fever gone is not typhoid gone. The relation of blood sugar to T2D is very similar. Bringing blood sugar back to normal is not an indication of diabetes gone.
There is an important difference in the two analogous situations though. We know the cause of typhoid quite well, and treatment that directly attacks the infective agent is available. Symptomatic treatment with antipyretics may be given as a supplement to the main anti-infective treatment but that is not the main course of the treatment strategy. This has not been so with T2D so far. Since we have not understood the underlying disease process as yet, conventional medicine has caricatured the symptom as the cause. Treatments to bring blood sugar to normal have been the focus of almost the entire research and clinical practice. This is partly a burden of history. Sugar was among the first markers of diabetes to be discovered. Therefore, all the thinking was stuck on to the only thing known. Now, since we know a lot more about the variety of changes in different systems of the body, the thinking needs to be reoriented. But we are still haunted by the ghost of history which makes us think that sugar is everything to diabetes. There is considerable amount of success in bringing the sugar down too, at least in the short run. But this approach does nothing to the roots of diabetes itself. It is not surprising that sugar control by drugs is also short lived and glycemic control worsens slowly.
Any theory of diabetes cannot be complete without explaining the changes in glucose homeostasis that take place in diabetes. I will argue in this chapter that the central nervous system is an important component of the mechanisms of glucose homeostasis and that without understanding the role of brain and behavior, we cannot understand glucose homeostasis. This argument is currently based more on theoretical and mathematical analysis. Currently experimental data are fragmentary but still sufficient to try and join dots to make a picture. The strength of the new model that I will describe in this chapter lies in its bold predictions that can account for most observed patterns that the orthodox paradigm is unable to account for.
More than 100 years ago, the celebrated physiologist Claude Bernard showed that puncture of the floor of the forth cerebral ventricle induced diabetes. This was a clear demonstration of the brain’s direct role in glucose homeostasis. But after the discovery of insulin, the role of brain was largely neglected. Although researchers are aware of it and there is revival of interest in possible central regulation of peripheral glucose [1], the picture is not yet sufficiently clear to bring about a substantial change in the prevalent thinking. Although the molecular mechanisms involved in signaling by glucose, insulin, and leptin in the brain are partially revealed, it is not yet clear in what way the signaling is impaired in diabetes and why.
At the level of clinical practice, brain’s role is practically denied, and in drug discovery, there are only feeble attempts to identify and use central drug targets for possible next-generation antidiabetic drugs. Any such attempts are faced with many problems so that a central action antidiabetic drug is unlikely to be in the market in the near future unless there is a radical change in the line of thinking [2]. There are some signs of change on the frontiers of research, but it has not gathered sufficient gravity, which is presumably due to the lack of appropriate paradigm rather than any methodological or implementational problems. The traditionally known circuit of glucose regulation on which the current clinical practice is based consists of the interplay between glucose, insulin, and glucagon. Raised plasma glucose induces insulin release by the pancreatic β cells, which facilitates glucose uptake by muscle and many other tissues. Insulin also arrests hepatic glucose production. This brings back the raised level of glucose. If glucose level goes below normal, glucagon release is stimulated which increases liver glucose production and thereby restores blood glucose levels. This sounds like a simple, nice, and efficient way of regulating glucose levels in blood.
But the reality is much more complex. The glucose–insulin–glucagon-dominated paradigm excludes or neglects a number of quite well-demonstrated phenomena. These include the role of parasympathetic inputs in insulin secretion, the cephalic phase of insulin response, direct vagus control of liver glucose production, the hepatic portal glucose sensors, the role of glucose-stimulated and glucose-sensitive neurons in the brain, and so on. By the orthodox view, the neuronal mechanisms are important only in the counterregulatory response to hypoglycemia and not otherwise [3, 4]. This is demonstrably not true. It has been shown, for example, that even for the “normal” regulatory function of insulin on liver glucose production, neuronal signaling is essential [5–7]. The orthodox view also fails to explain a number of “inconvenient” findings that we discussed in Chap. 3 which I will briefly reiterate now because any new theory needs to explain most or all the odd facts. The questions that the new theory needs to answer are as follows: (1) Why in an HIIR state suppressing insulin production does not lead to rise in plasma glucose, instead the insulin sensitivity increases? (2) Why in MIRKO mice high muscle insulin resistance and lack of compensatory hyperinsulinemia fail to increase blood sugar? (3) Why in LIRKO mice in spite of liver insulin resistance the fasting blood glucose spontaneously returns to normal but in diabetes it does not? (4) Why perfusion of glucose directly into the brain results in peripheral hypoglycemia? or (5) Why in some patients plasma glucose fails to respond to insulin, and further why infusion of insulin paradoxically increases blood sugar levels in some patients (Somogyi paradox)? (see Chap. 3 for details of all these paradoxes). We need a model of glucose homeostasis that accommodates all the known sensors of glucose levels in the body and the brain and all known effectors of glucose dynamics in such a way that the so far unexplained patterns in glucose homeostasis get adequate explanation. Of particular importance are the changes in the glucose and insulin curves during GTT. There are four different features of altered GTT curve in early diabetes: (1) delayed insulin response, (2) increased height of the glucose peak from the fasting glucose level, (3) increased fasting glucose level, and (4) a long right-hand tail, that is, a delay in returning the glucose level back to fasting level. Any theory of diabetes should explain all the four together. It should also explain differential occurrence of some of them, that is, IGT in the absence of IFG and vice versa. Any new theory needs to show how it explains these features of the altered curve better than the classical theory. The new model also needs to either find support in the existing literature or make experimentally testable predictions. Let us try our luck to find such a theory of diabetic hyperglycemia now.
The mechanisms involved in glucose sensing can be grossly classified in two categories, namely, (1) peripheral, consisting of direct effects of glucose on the α and β cells of the islet, and (2) central or neuronal, consisting of glucose transport to the brain, glucose utilization by the brain, glucose sensing neurons, information processing, neuronal signals to pancreas to regulate insulin and glucagon production, and direct neuronal signals to liver to regulate glucose production. If two parallel circuits of homeostasis exist in the body, we can theoretically construct four possible models of glucose homeostasis: (1) one in which only the peripheral mechanisms work, which is the traditional model, (2) one in which only the central mechanisms work, (3) one in which both work together all the time, and (4) one in which the two operate conditionally, that is, under one set of conditions only the peripheral control works and in other set of conditions the central mechanisms take over the control. A detailed formal mathematical model considering the possible outcomes of all the four combinations is described in Appendix IV. Here it is represented by a schematic diagram (Fig. 12.1), and we will discuss in this chapter the intuitive outcomes of the model alone without worrying much about the mathematics.
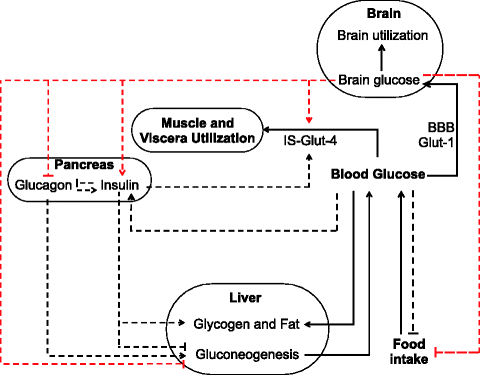
Fig. 12.1
A schematic diagram depicting the peripheral and central regulation of glucose homeostasis (IS insulin sensitivity, BBB blood–brain barrier). Solid black lines depict glucose flow, black dotted lines peripheral regulation signals, red dotted lines central regulation signals
It is not difficult to understand that a peripheral mechanism alone would be more efficient in maintaining plasma glucose level and bringing it back after any perturbation. This is because peripheral control has less number of steps in the feedback loop and therefore minimum delay between sensing a change and mobilizing effectors in response to the change. A mechanism that involves central sensing of brain glucose levels has more number of steps that are likely to cause a delay in response. Glucose in plasma is transported to the brain through specific transporters in the blood–brain barrier called glut-1 [8]. The brain level of glucose or the rate of energy production in the brain is sensed by the glucose sensing neurons; they send signals to the arcuate nucleus of the hypothalamus, where the information is processed and neuronal signals are given to the pancreas to secrete insulin or glucagon and also directly to the liver to regulate glucose production [9]. The role of neuronal signals in the function of endocrine pancreas is long known, but its importance is being realized only recently [10–13]. The neuronal steps in this process are rapid and therefore will not cause any significant delay in the circuit. However the single process that can cause substantial delay is the transport or diffusion of glucose from plasma to the brain. So if brain glucose is being sensed instead of plasma glucose, it will result in some delays in regulation. It is well known in cybernetics that a delayed feedback causes oscillations. It is not surprising therefore that peripheral control is more efficient than central control in minimizing deviation in plasma glucose under any positive or negative peripheral perturbation such as food intake or muscle exercise. We can perceive three types of perturbations in steady-state plasma glucose which are commonly experienced in everyday life. One is energy intake which transiently increases plasma glucose, the other is muscle exercise that causes rapid uptake of plasma glucose by muscle, and the third is increased brain demand that can be triggered by intensive mental activity or mental “stress.” The stress can be acute or chronic. In all these types of perturbations, peripheral control is more efficient in maintaining stable plasma glucose and returning to it quickly after the perturbation.
Why does the central control exist then? The prevalent thinking is that the central control is an emergency measure that would prevent brain damage if glucose level in the brain suddenly reduces. This is called the counterregulatory response [14, 15]. The current perception is that the counterresponse operates in case of severe hypoglycemia, that is, if the brain glucose levels dangerously reduce as a result of reduction in plasma glucose levels. This, however, does not make much sense from the system design point of view. If a drop in brain glucose levels is caused by a drop in plasma glucose levels since that is the only source of glucose for the brain, sensing a drop in plasma glucose level would work more efficiently than waiting for the brain level to drop and then act. For a sensitive and important organ like the brain, if it was to be protected against hypoglycemia, sensing plasma levels would have been the best strategy. Hence peripheral glucose sensing would have been more efficient even for a counterregulatory response to hypoglycemia. But surprisingly no neuronal mechanism of sensing plasma glucose appears to have evolved. Neuronal sensing of glucose happens only in the brain. Therefore either the system is badly designed or it is evolved for a purpose other than protecting the brain from hypoglycemia.
There is an alternative scenario when sensing brain glucose would be more efficient than sensing plasma glucose and that is if the demand for glucose in brain tissue goes up acutely or chronically. The steady-state concentration of glucose in the brain, in the absence of perturbations, is decided by the rate of glucose diffusion from blood and the rate of consumption by the brain tissue. The brain consumes disproportionately high quantities of glucose. Although the total weight of the brain is only about 2–3% of the body, it consumes about 20% of total body glucose at rest [16]. If glucose consumption by brain tissue increases, the steady-state glucose level in the brain will fall. The only means to correct it is by increasing transport from plasma since the only source of glucose to the brain is plasma. If brain glucose levels are depleted by explosive brain activity, the primary change is in the brain and not in the plasma, and therefore, sensing brain sugar would be specifically needed and sensing plasma sugar would be useless. Therefore, from a system design point of view, we can argue that the central mechanisms of glucose regulation must have evolved not to protect brain from a drop in plasma glucose but to ensure adequate glucose supply under conditions of increased brain demand. In other words, if the cause of brain glycopenia is hypoglycemia, then central mechanisms would be highly inefficient in dealing with the situation. However, if the cause of neuroglycopenia lies within the brain, central mechanisms would be highly efficient in bringing glucose supply of the brain back to normal. Therefore it would be logical to assume that the central mechanism evolved primarily to handle neuroglycopenia caused by explosive brain activity. But the same mechanism may also work to protect the brain from hypoglycemia, although it will not be too efficient in this task.
When do we expect a higher glucose demand in the brain? This can happen when facing acute or chronic environmental or social challenges that need computation of a complex solution. Such a rise in brain glucose demand has been demonstrated [17, 18]. Here we return to our soldier–diplomat dichotomy. Based on the soldier–diplomat hypothesis, I am going to make a rather bold speculation here. On adopting a diplomat lifestyle over soldier, not only the brain glucose demand increases, the brain glucose levels become more important than plasma glucose levels, and the primary focus of glucose homeostasis shifts from maintaining plasma glucose levels to maintaining brain glucose levels. The idea that the true target for glucose homeostasis is not plasma glucose but brain glucose is not new [19]. Over the last decade, a group of researchers have been promoting the concept of “selfish brain” that exerts an active energy pull. According to this theory, glucose homeostasis, and particularly glucose supply to the brain, is not governed by a push from glucose availability but rather an active pull from the brain [20–26]. The proponents of the selfish brain theory postulate further that a healthy state is marked by a “competent brain pull,” whereas an “incompetent brain pull” leads to obesity and diabetes.
While I am happy with the idea of a brain pull and in some sense the central regulation used in the analysis below can be taken to be equivalent to a brain pull, I differ from these ideas in proposing that there is flexibility in the homeostasis priority. In soldier life, muscle glucose supply for explosive muscle activity is a priority, whereas in diplomat life, long-term sustained supply to the brain is the priority. This shift in priority is expected to be adaptive because the nature of challenge for glucose homeostasis in the two lifestyles is likely to be different. In soldier life, explosive muscle respiration is the most frequently expected perturbation. Brain activity is certainly involved in complex nerve muscle coordination during a physical encounter, but this is typically of a short duration. On the other hand, muscle building and maintenance are needed over sustained time scale. In contrast, in diplomat life, intensive brain activity may be more frequent and prolonged. Simulations show that when intensive muscle glucose utilization perturbs glucose homeostasis, peripheral control is highly efficient in restoring normality of plasma glucose. Central control is less efficient. On the contrary, if the primary challenge consists of increased brain glucose utilization, then central control is more efficient in restoring brain glucose homeostasis at the cost of raised plasma glucose (Fig. 12.2).
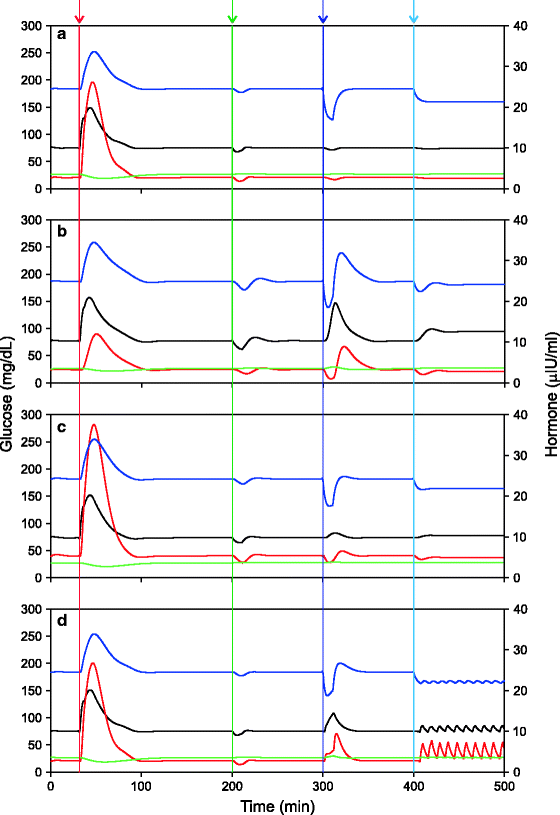
Fig. 12.2
Four possible ways in which peripheral and central regulation of glucose may interact and the relative performance of the four systems against four different perturbations: (a) peripheral control alone, (b) central control alone, (c) joint central and peripheral control with x = 0.5 (see Appendix IV), and (d) switchover control where central mechanisms take over if brain glucose levels reduce below a threshold (22 mg/dL in these simulations). Four types of perturbations were introduced at four time points shown by vertical lines from left to right respectively: food intake, acute muscle exercise, acute brain exercise, and sustained brain exercise. It can be seen that peripheral control is most efficient in maintaining stable plasma glucose with all possible types of perturbations, but it allows the brain glucose to reduce to much lower levels during sustained brain hyperactivity. The central control is not very efficient in maintaining plasma glucose but maintains stable brain glucose particularly during sustained brain hyperactivity. The switchover control is good at handling acute brain exercise but may lead to rapid oscillations under sustained increase in brain activity. The oscillations are caused by alternately switching to central control when brain glucose goes below the threshold and switching back to peripheral when it returns to normal. Black line: plasma glucose, red line: plasma insulin, green line plasma glucagon and blue line (secondary axis) brain glucose. The same color code is used in figures 12.8, 12.9, 12.11 and 12.13
The suggestion therefore is that in a diplomat lifestyle the central mechanisms of glucose homeostasis should be more active than the peripheral ones. This central shift is of quantitative nature on a continuous scale which can be mediated by a change in the sensitivity of pancreas and liver to peripheral versus central signals. In soldier life, islets should be more responsive to peripheral glucose stimulation than to autonomic stimulation, and in diplomat life, autonomic activation should be more important for insulin secretion than stimulation by peripheral glucose. Similarly liver glucose production should be more under feedback control directly by glucose and insulin in soldier life and more under neuronal control in diplomat life. This idea is certainly testable, and there is already some evidence in this direction. Pancreatic islets are richly innervated by parasympathetic and sympathetic neurons [27]. These play some role in the regulation of insulin secretion under normal healthy state and under all conditions including meal-stimulated insulin release [28] or exercise-induced insulin suppression [29]. The autonomic system is involved in determining the glucose sensitivity of β cells [30]. Vagotomy can partially abolish the altered responsiveness of β cells exposed to chronic hyperglycemia [30]. Our hypothesis expects that in the obese, insulin-resistant, prediabetic, or early diabetic state insulin secretion should become less responsive to peripheral glucose and more responsive to central stimuli. Such a shift in response is demonstrated by the comparative response to glucose versus that to the cholinergic agonist carbachol. In fat-fed C57BL/6J strain of mice, after 12 weeks of fat feeding the glucose-induced insulin secretion was markedly impaired but carbachol-stimulated insulin secretion was potentiated [31]. This indicates a possible shift in the responsiveness of β cells from peripheral to central stimuli and more generally a change in emphasis of the system from peripheral to central mechanisms. In a human study, cholinergic enhancement of insulin secretion was stronger in obese than in non-obese subjects [32], indicating that obesity is associated with increased sensitivity of β cells to autonomic signals. In diabetics without autonomic neuropathy, the basal and meal-stimulated levels of pancreatic polypeptide are higher than normal [33, 34]. The pancreatic polypeptide is a marker of autonomic activity in the pancreas, and therefore, higher levels of this peptide in diabetes suggest greater role of autonomic inputs in pancreatic functions. It has been suggested that stress-related suppression of insulin is actively mediated by the brain [22], and this is likely to happen through autonomic control. In diabetic rats, β cells showed enhanced sensitivity to sympathetic signals [35]. The liver cell membranes of ob/ob mice have threefold higher β adrenergic receptor binding sites and show threefold increase in response to catecholamines than controls [36]. This again indicates that liver and pancreatic β cells might be becoming more sensitive to central regulation, along with losing sensitivity to peripheral insulin and glucose signals, respectively. The demonstration that the increased response of liver cells to central stimuli is mediated by corticosteroids [36] raises the possibility that HPA might be an important mediator of the central shift. Since HPA is a marker of diplomat life (Chap. 11), our notion of a central shift in diplomat life is supported.
PPAR gamma coactivator-1(PGC-1) is an important modulator of gluconeogenesis in the liver [37, 38], and PGC-1 expression is shown to be under sympathetic influence [39, 40]. Interestingly PGC-1 expression is downregulated in muscle and adipose tissue where its downregulated state is correlated to insulin resistance [41–44] and it is upregulated in liver where its upregulation correlates with liver insulin resistance [37, 38]. This paradox (one of the several dozen associated with insulin resistance) can be resolved by the differential action of the sympathetic nervous system. In a nonaggressive diplomat lifestyle, there is lowered sympathetic input to muscle and adipose tissue. According to the above argument, it should be increased in the liver. In diplomat life, visceral fat lipolysis and muscle activity are downregulated. At the same time, sympathetically stimulated liver glucose production is upregulated. Both seem to be achieved through mediation by PGC-1 but in opposite directions. This is a nice demonstration of how the sympathetic system through the agency of PGC-1 mediates fine-level changes in muscle, fat, and liver. All the changes are in different directions but bring about a consorted and organized action. All this is more than preliminary evidence in support of my speculation that diplomat life is characterized by increased importance of autonomically mediated central regulation of glucose homeostasis and decreased importance of peripheral regulation.
It is necessary to clarify here the relationship of this central shift with counterregulatory response. If we visualize a zero to one scale, zero represents complete peripheral control as visualized by the current working clinical model. The argument above states that in a soldier life, the system may be say at a score of 0.2 or 0.3, and on adopting a diplomat life, it may gradually shift to say 0.6 or 0.7. The all-out counterregulatory response stands at 1. This zero to one conceptual scale is incorporated explicitly in the model in the parameter x (Appendix IV). Thus, even after a soldier to diplomat transition, a counterregulatory response is possible, but the more the shift, the less will be the difference between the basal standing state and the all-out counterregulatory response. This will reflect into an apparently blunted counterregulatory response in diabetes.
There are two alternative ways in which the central mechanisms influence plasma glucose. One way is by modulating pancreatic hormones insulin and glucagon and the other by direct neuronal modulation of liver glucose production. If glucose utilization by the brain is increased owing to increased cognitive demands, higher level of plasma glucose is needed. This can be achieved by suppressing insulin and enhancing glucagon production. But suppressing insulin may not be desired since insulin itself is needed for cognitive functions. Therefore, the other pathway, that of direct neuronal regulation of liver glucose production, could have evolved. With two alternative pathways available within the central regulation mechanisms, it becomes possible to fine-tune glucose and insulin levels independently. If the involvement of insulin was mandatory in the regulation of glucose, suppression of insulin would have been crucial to increase brain supply of glucose. However, since the cognitively active brain needs both glucose and insulin simultaneously, stimulation of insulin production along with suppression of insulin action on the liver and direct regulation of glucose production by autonomic control would be a better strategy. This will result in a high fasting HOMA-IR index, although there is nothing wrong with the liver. On the other hand, whenever cognitive demand for insulin is not very high but liver glucose production is needed (such as during starvation [45]), suppression of insulin and stimulation of glucagon production might be a more desirable pathway. The presence of these two alternative central pathways to regulate glucose production brings this flexibility, and the brain must be smart enough to make contextually differential use of them.
For a cognitively intensive diplomat life, direct neuronal regulation of liver glucose production would be more important than regulating pancreatic hormones. There is increasing evidence that hypothalamic neuronal signals through the vagus nerve play an important role in regulating liver glucose production [46–49]. Moreover, it is also possible that the “direct” action of insulin on liver glucose production is not really “direct” but depends upon its interaction with neuronal inputs [50]. This hypothesis expects that a direct neuronal control over liver glucose production or contribution of neuronal mechanisms in its regulation will be more important in a diplomat than in a soldier personality. Since the neuronal regulation of liver glucose production is under sympathetic control, there would be increased sympathetic activity in a diplomat’s glucose homeostasis. Glucose levels in the brain appear to be finely monitored and modulated depending upon the activity and respiration rate of brain tissue [51–54]. In effect, in a diplomat lifestyle, there could be activation of mechanisms that modulate the plasma glucose in such a way to maintain steady and adequate brain glucose level.
It is possible that the central effects on pancreas and liver take two different time courses. One consists of active and ongoing neuronal modulation of insulin production and insulin action on the liver. The other could be long-term programming of liver and β cells. The latter has a possible adaptive value. For a chronically altered behavior, a chronic change in pancreatic and hepatic response is likely to be more economical than ongoing active modulation by neurons. Further, if the peripheral system is kept active, there can be conflict of interests. For example, if the brain needs chronically elevated plasma sugar, it is necessary to reduce β cell responsiveness to glucose. Otherwise, the central and peripheral systems will work against each other and waste much energy in the conflict. Similarly, along with neuronal regulation of liver glucose production, it is necessary to reduce insulin sensitivity of the liver; otherwise, the two systems will be in conflict. The suggestion logically emerging from this is that the loss of β cell responsiveness and liver insulin resistance could be strategies to reduce the conflict between the two homeostatic systems. It is also likely that β cells are programmed neuronally [30] on a prolonged exposure to hyperglycemia to reduce insulin production since chronic hyperglycemia could be a marker of altered homeostatic strategies. In effect, the new hypothesis states that the diplomat behavioral strategy is supported by a shift in the glucose homeostatic mechanisms from peripheral to the central ones which is actively mediated by the autonomic nervous system.
One can clearly appreciate at this level that a central shift in homeostatic mechanisms combined with increased brain glucose demand is sufficient to give rise to hyperglycemia along with reduced sensitivity of β cells to glucose and reduced sensitivity of liver to insulin. This can happen without β cell degeneration. This is a strong candidate competing with IR-RII for explaining hyperglycemia in diabetes. But this is not the complete story. There are more players in the act of central regulation.
Vagus control of hepatic function not only regulates glucose production by liver but also fine-tunes muscle insulin sensitivity. Interruption of the hepatic parasympathetic nerves by surgical denervation, atropine, or blockade of hepatic NO synthase induces peripheral insulin resistance [55]. In a response to the synergistic action of insulin, parasympathetic signal, and hepatic NO production, a hormone called hepatic insulin sensitizing substance (HISS) is produced which increases postmeal insulin sensitivity of specifically muscle tissue [55, 56]. This means that the central system is also capable of manipulating muscle insulin resistance. Muscle insulin action has HISS-dependent and HISS-independent components, and the HISS-dependent component is responsible for about 56% of muscle glucose uptake. The proportion of insulin action under central control is substantially large to affect peripheral glucose dynamics.
Another important player influencing central regulation is the glucose transporter in brain capillaries called glut-1. An alternative possible way of increasing the glucose supply of the brain would be to increase the amounts of glut-1 across the blood–brain barrier (BBB). Glut-1 action is insulin independent unlike the muscle glucose transporter glut-4. We have seen earlier that muscle insulin resistance is a mechanism by which investment in muscle is decreased by reducing glut-4 activity. By the same logic, if the investment in brain is to be increased, increasing glut-1 activity would be appropriate. Since glut-1 is insulin independent, the only way this can be done is by increasing the levels of glut-1 itself.
However, there is a serious problem here. The plasma glucose levels are subject to wide fluctuations normally caused by food assimilation after meals, muscle uptake following exercise, and other physiological perturbations. The brain glucose levels are relatively constant, and glut-1 has an important role in maintaining steady glucose levels in the brain. It can be shown mathematically that the lower the levels of glut-1, the steadier will be the brain glucose levels. Simulations (Fig. 12.3) showing the effect of glut-1 levels on the ratio of the amplitude of fluctuations in plasma and brain illustrate this. The principle can be intuitively appreciated through a cartoon (Fig. 12.4) which helps us imagine a high-amplitude wave being forcibly pushed through a narrow slit so that the wave coming out on the other end has compressed amplitude. If a stable glucose supply to the brain is desired in diplomat life, then to protect the brain from fluctuations in plasma glucose, a decrease rather than an increase in glut-1 will help. However, a decreased glut-1 will also result in a decreased mean brain glucose level. The solution to this problem is to bring about a compensatory increase in the plasma glucose. This is possible only if the central regulation is in operation. The model shows that rather than increasing glut-1 levels, increasing mean plasma glucose and slightly reducing glut-1 ensure a more steady supply of glucose to the brain (Fig. 12.3). A moderate reduction in glut-1 can therefore be adaptive for a diplomat lifestyle in coordination with central regulation mechanisms. However, a supernormal reduction in glut-1 can be detrimental as shown by these simulations.
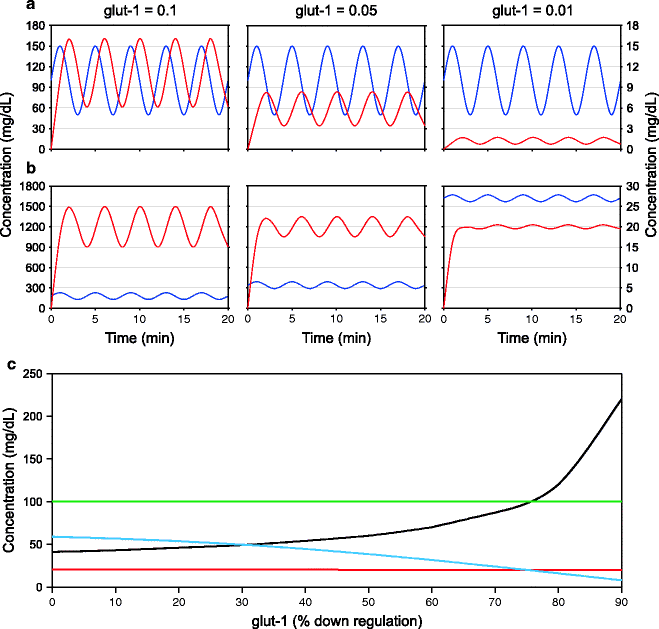

Fig. 12.3
Effect of the width of glut-1 window on stability of blood glucose levels: Here, in a model system, plasma glucose (blue line, primary Y axis) is given an oscillation of constant amplitude in a sine curve. The resulting oscillations in brain glucose are monitored (red line secondary Y axis). (a) As glut 1 decreases from 0.1 to 0.01 (arbitrary units), the amplitude of oscillation of brain sugar decreases but so does the mean. (b) If the mean brain glucose is to be kept constant and the amplitude is to be decreased, decreasing glut-1 and increasing mean plasma glucose are the only possible solution. (c) The behavior of mean and amplitude of plasma and brain glucose with different levels of glut-1. In these simulations, the amplitude of plasma sugar oscillations is kept constant (green line). The mean plasma glucose (black line) is adjusted to keep a constant mean brain glucose (red line). With greater suppression of glut-1, the amplitude of fluctuation of brain glucose decreases (blue line), but the mean plasma glucose needed to achieve this increases disproportionately. This is what is expected if brain glucose stability is the main homeostatic target, and there is reduction in glut-1 levels
Fig. 12.4
A small glut-1 window can effectively dampen fluctuations in glucose concentration
Now, along with central shift of the homeostatic mechanisms and increased brain glucose demand, glut-1 downregulation is an additional factor that can contribute to raising plasma glucose. This logic raises the possibility that it is the brain glucose dynamics that primarily underlies diabetic hyperglycemia and IR-RII is incidental. This argument is elaborated more with some mathematics in Appendix IV, and readers that are not averse to mathematics and expect more rigorous argument may visit Appendix IV. For mathematics-averse readers, the set of arguments above should be sufficient to convince that at least a logical possibility of alternative reason for diabetic hyperglycemia exists and needs serious investigation.
The levels of glut-1 in brain capillaries indeed go down in diabetes, but the data are fragmentary. There are no studies on glut-1 levels across the brain capillaries in human diabetes to the best of my knowledge. In rat models of diabetes, glut-1 levels have been repeatedly shown to reduce, but the experiments lack consistency [57] presumably due to methodological problems and therefore are somewhat difficult to interpret. There can be several reasons for the inconsistency across experiments. The nonlinearity of glucose transport, difference between different models of diabetes, methods of estimating glut-1 levels or glucose transport rates, failure to make observations at steady-state concentrations, need for sufficiently long period for detection of a chronic response, regional differences in glucose dynamics [58, 59], and a number of other factors need to be taken care of in such experiments. In spite of these problems, a general pattern emerging from many experiments is that glut-1 levels are downregulated in response to hyperglycemia and upregulated in response to hypoglycemia [8, 60–68]. As perceived currently, glut-1 reduction is a consequence rather than the cause of hyperglycemia. Another general and robust trend in all the studies appears to be that the alterations in glut-1 levels are slow. Chronic hyperglycemia, at least over several weeks, is needed for detectable downregulation of glut-1. Upregulation by hypoglycemia could be a little faster but still requires several days to show detectable effects.
Several questions are raised by the demonstration of altered glut-1 levels in diabetic rat models, and I consider two of them to be of prime importance. The first is what causes reduction in glut-1 and why. The second is what the implications of reduced glut-1 are for glucose homeostasis. We will look at the latter first since that emphasizes the importance of altered glut-1 levels. Once we know how important it is, the pursuit of the first question will get more weighting. When we start examining the possible consequences of reduced glut-1, we find explanations for a number of observed patterns in diabetes. This leads to a strong alternative reasoning for diabetic hyperglycemia along with a number of characteristic changes associated with it.
1.
Raised fasting plasma glucose: The lower the glut-1 expression, the slower will be the rate of glucose transport across the blood–brain barrier (BBB). This would lead to a lower steady-state brain glucose level if the rate of uptake by brain tissue is assumed constant. However, if a desired steady-state level has to be maintained in spite of reduced glut-1, the only way is to increase plasma sugar. This can happen only when the central regulation is sufficiently active, that is, x of the model is sufficiently large. The lower the levels of glut-1, the greater would be the plasma glucose needed to maintain a given level in the brain. Therefore, reduced glut-1 can be a cause of hyperglycemia. If this level falls, brain glucose level will fall, and the autonomic control will increase blood sugar by activating liver glucose production. For this to happen, any defect in β cell is unnecessary. The lower level of brain glucose as sensed by glucose sensing neurons will suppress insulin production in spite of β cells being normal and/or increase liver glucose production in spite of liver cells being normal.
2.
Failure of adjustment of insulin sensitivity to decreased insulin production: We have seen in Chap. 3 that in an HIIR state, if insulin production is artificially suppressed, instead of glucose level rising, insulin sensitivity increases such that plasma glucose remains normal. This well-demonstrated phenomenon is not taken into account by the IR-RII paradigm. Experiments demonstrate the existence of a mechanism which brings about compensatory increase in insulin sensitivity when insulin production is suppressed. If a compensatory rise in insulin sensitivity is possible in experimental suppression of insulin production, what prevents it from happening in diabetic hyperglycemia? Lowered level of glut-1 is a promising answer to this question. According to this hypothesis, the transition from insulin-resistant to hyperglycemic state is not caused by β cell dysfunction but by altered brain glucose dynamics. Under central shift and reduced glut-1 state, it is in the interest of the brain to raise plasma glucose, and therefore, central signals reduce insulin secretion and/or enhance liver glucose production so that a higher plasma glucose level is maintained. Here, the primary cause is not insulin resistance, and therefore, even if insulin sensitivity is restored following insulin suppression, it cannot prevent hyperglycemia.
3.
Beta cell dysfunction? In this model, the central system decides what the plasma glucose level should be to attain the desired steady-state brain glucose. Insulin and glucagon secretions and sympathetic stimulation of liver for glucose production are neuronally regulated to achieve these levels. When neuronal control takes on a predominant role, insulin secretion is no more directly responsive to glucose, independent of neuronal inputs. If brain needs increased supply of glucose, it will suppress insulin production and/or enhance glucagon secretion and liver glucose production. As a result, β cells will produce less amount of insulin than what is expected from them at the standing glucose level. This could give an impression of β cell dysfunction when nothing is actually wrong with them. Researchers unaware of neuronal control or choosing to ignore it are bound to think that as β cells are not responding to glucose as expected, there must be some kind of functional β cell defect. Further, if insulin production is chronically suppressed by the neuronal system, disuse atrophy may slowly reduce the β cell mass. Chronic suppression of insulin secretion should be accompanied by chronic suppression of amylin secretion too since both are cosecreted. But amylin has much longer half-life than insulin, and amylin disposal is presumably more difficult than insulin. As a result, excess amylin that is not secreted in time could slowly accumulate and form amyloid deposits in the islet tissue. The amyloid deposits which are long known to be associated with islet degeneration [69–72] could be a consequence rather than a cause of reduction in β cell secretary activity. Alternatively, once amyloid deposition begins, islet degeneration may begin.
4.
Diminished AIR and delayed insulin response in GTT: After a meal or oral glucose intake, there is a rapid and short-lived burst of insulin production which is called acute-phase insulin response (AIR). In diabetes AIR is suppressed and even undetectable. This is often suspected to be the prime reason of impaired GTT. We have seen before (see Chap. 3) that neuronal inputs have a major role in AIR [73–77]. Even in ivGTT, there is a neuronal component in AIR, although it may be less important than in oral GTT [78, 79]. For ivGTT, neuronal inputs presumably come when the glucose-responsive neurons in the brain respond to the rising concentration of glucose in the brain. With reduced glut-1 and thereby reduced rate of diffusion to the brain, the glucose-responsive neurons may respond with a delay, or if they require some threshold rate of rise in glucose concentration for AIR which is not achieved with very low levels of glut-1, AIR may be completely absent. Currently, the blame for the absence of AIR is placed on β cell “defect,” but the nature of the defect or mechanism by which β cells respond with a delay is not known. Glut-1 reduction offers an alternative hypothesis for this phenomenon which appears more logical, and the possibility needs to be tested experimentally.
In IGT and early diabetics, the total amount of insulin produced by the pancreas is not reduced; in fact, it may have increased substantially. But there is a delay in the beginning of the insulin response. This is likely to be an effect of central shift and glut-1 reduction which delays the rise in the levels of brain glucose (Fig. 12.5). Assuming that neuronal inputs become more important for insulin release in a diplomat state, glut-1 downregulation rather than β cell defect is a better model for explaining the delayed insulin curve. Since the total amount of insulin produced is not reduced, β cell capacity to produce insulin cannot be doubted. Again, what kind of “defect” makes the β cells work with a delay but without loss of capacity is not known. In in vitro experiments, altered responsivity of β cells to glucose has been shown, but I failed to find an experiment demonstrating the phenomenon of delayed response. This suggests that delayed response is not a property of β cells. Therefore we need to examine glut-1 reduction and central shift as an alternative hypothesis for the altered AIR and GTT.
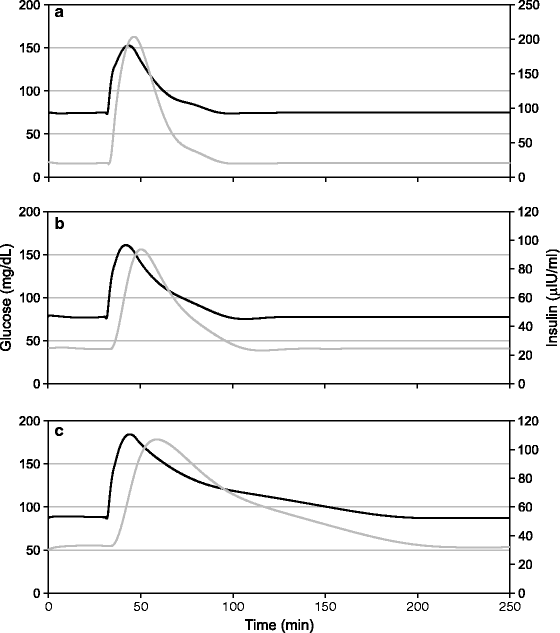
Fig. 12.5
Shift to central regulation and glut-1 reduction causes delay in insulin response to glucose. (a) OGTT with peripheral control, (b) OGTT with central control, and (c) OGTT with central control and reduced glut-1. Note the progressively rightward shift in the insulin peak. Only models involving central control lead to delayed insulin response. The peripheral control model does not give this feature of altered OGTT under any parameter values
5.
Triggering counterregulatory response at higher plasma glucose: If there is glut-1 reduction, there would be lower levels of brain glucose per unit plasma glucose at steady state. Since the glucose sensing mechanisms in the brain sense the brain glucose levels and do not have a direct access to plasma levels, the counterregulatory response may be triggered at a higher plasma glucose level which is known to happen in diabetics [80–82].
6.
Why the counterregulatory response in diabetics is delayed and less glucagon dependent: It is known that in long-term diabetics, the glucagon response to hypoglycemia is blunted [83]. Since the rate of diffusion from blood to brain is reduced because of reduced glut-1, a sudden drop in blood sugar will also be sensed more slowly by the brain and the counterregulatory response will be delayed. This is compatible with the observations [84]. It is possible that as a compensatory measure against the delay, the hormonal glucagon response which is independent of sympathetic activation [85] is bypassed and replaced by direct autonomic stimulation of liver glucose production [86] which is a faster response. The delay in sensing could thus be partially compensated by reduction in response time.
7.
Shift in baseline autonomic tones: Both sympathetic and parasympathetic systems have different roles in glucose homeostasis. Sympathetic activation suppresses insulin and enhances liver glucose production. Parasympathetic on the other hand enhances insulin production and also, through the agency of HISS, facilitates muscle glucose uptake. Logically, if the brain is short of glucose, the sympathetic system should be activated, and if there is excess glucose, parasympathetically activated insulin production and diverting glucose to muscle would help in regulating brain sugar. If along with diabetic hyperglycemia brain sugar also had gone up, we would expect parasympathetic activation. On the contrary, sympathetic activity predominates, and parasympathetic is suppressed in diabetes. This pattern gets an explanation if glucose transport to the brain is assumed to be reduced by reduced glut-1, reduced capillary blood flow, or increased glucose consumption in the brain.
8.
Rise in stress-related sugar: When the glucose demand of the brain goes up which may happen in emotional arousal, worries, or anxiety, the increased demand can be met by slightly increasing the plasma glucose levels. Figure 12.6 demonstrates the interesting dynamics of this under high and low levels of glut-1. The hypothetical diagram is not based on data since data on brain glut-1 levels in humans under different physiological states is conspicuously absent, but it illustrates conceptually what may be happening with glut-1 reduction. Glucose transporters operate by facilitated diffusion and therefore follow Michaelis–Menten kinetics. At high level of the transporter, an increased demand can be met with by a small rise in plasma glucose since in this region, the slope of the line is steep. A reduction in glut-1 changes the nature of the curve such that the slope of the line in this region is much smaller, and as a result, a large change in plasma glucose is needed to fulfill a small increase in brain demand. This is an alternative explanation for stress- or anxiety-induced rise in plasma sugar. Again, there is a need to test this model in comparison with the conventional model of cortisol-mediated rise in sugar. The two are not mutually exclusive and may work in harmony.
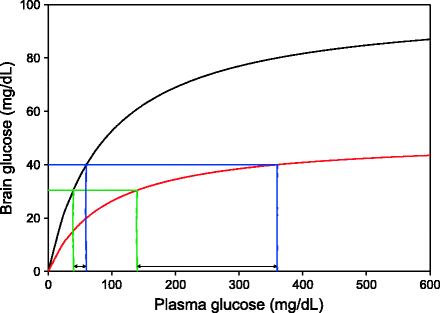
Fig. 12.6
Sugar response to mental stress at two different levels of glut-1: The dynamics of glut-1-mediated glucose transport follows saturation kinetics. If we assume that the normal glucose requirement of the brain (green line) is fulfilled by some steady-state plasma glucose levels, a slight increase in brain glucose utilization (blue line) necessitates a marginal increase in plasma glucose if glut-1 level is high. The same requirement puts greater demand on plasma glucose if glut-1 is reduced (red curve)
9.
Why HOMA-based and other indices may not reflect insulin resistance in advanced T2D: There are a large number of indices for measuring peripheral insulin resistance. In normoglycemic conditions, most of them correlate quite well with the more elaborate “gold standard” measurement of insulin-mediated glucose uptake, namely, the euglycemic hyperinsulinemic clamp. The correlations become substantially weaker as diabetes advances [87–91]. My interpretation of this phenomenon is that as diabetes advances brain glucose dynamics and central regulation become increasingly important in determining blood glucose. Direct neuronal action rather than glucose feedback insulin action decides fasting glucose levels in advanced stages. Since the causal role of insulin and insulin sensitivity in deciding plasma glucose is substantially undermined, the correlations become weak. Central shift and glut-1 reduction can increase HOMA-IR without increasing insulin resistance because of which HOMA-IR does not faithfully reflect insulin resistance.
10.
Making exogenous insulin ineffective: The model shows that even if hyperglycemia is caused by glut-1 reduction, it may still respond to exogenous insulin as long as the shift in emphasis from peripheral to central regulation is moderate. It is likely that in the supernormal diplomat lifestyle of modern society, there is a supernormal shift towards central regulation. If this happens, clinically feasible doses of exogenous insulin will be unable to bring glucose to normal levels. This indeed happens in many advanced diabetics for whom normal glucose levels are not attained even after exceedingly high doses of insulin. Going by traditional thinking, these patients must have extremely high insulin resistance. But this is not supported by empirical findings. MIRKO mice have demonstrated that muscle insulin resistance does not raise fasting plasma glucose levels even if there is no compensatory rise in insulin [92]. In human studies, it is estimated that peripheral insulin resistance reaches a plateau at FPG levels of 160–180 mg/dL and does not contribute to further rise in glucose. Thus there is a limit to insulin resistance and its effects on the rise in blood glucose level. Any rise beyond this is mainly contributed by excessive liver glucose production rather than reduced glucose uptake [93]. But liver-specific insulin receptor knockouts have demonstrated that even after complete liver insulin resistance, fasting sugar increases moderately and returns to normal in a few months. Restoration of normoglycemia is because of central regulation compensating for liver insulin resistance independent of insulin action [94, 95]. This means that neither muscle nor liver insulin resistance can account for extreme hyperglycemia not responding to insulin. If the picture is upside down, that is, increased glucose production is caused by central mechanisms, independent of insulin, exogenous insulin is unlikely to have much effect on it.
Further it is known for a long time that in a class of patients, large doses of insulin paradoxically increase hyperglycemia which is ameliorated only after reducing insulin dose (Somogyi phenomenon) [96]. This appears highly counterintuitive. It was initially suspected that large doses of insulin first lead to hypoglycemia, and then the counterregulatory response increases plasma glucose. However, continuous monitoring of glucose in such patients has failed to detect a hypoglycemic phase [97]. This paradox is best explained by the glut-1 model and by the glut-1 model alone. In patients with very low glut-1, the difference in plasma and brain glucose levels is large, and as a result, a high dose of insulin reduces brain glucose levels below the threshold even when the plasma levels are above normal. In effect, insulin stimulates the counterregulatory response even without hypoglycemia. Withdrawing insulin in such patients improves glycemic control.
11.
Glucose infusion in the brain reduces plasma glucose: Perhaps the most direct evidence of how brain glucose dynamics affects plasma glucose levels comes from the experiments that infused glucose in the intracerebroventricular space [98]. This infusion of small quantity of glucose induced a rapid reduction in plasma glucose, indicating that plasma glucose is controlled by the brain glucose sensing and regulating mechanisms even under normoglycemic conditions. Another important inference of this experiment is that this mechanism works both ways. It is well accepted that lowering brain glucose triggers the counterregulatory sympathetic mechanisms. But the perception has mostly been that this is only an emergency-triggered mechanism which otherwise remains silent. If increasing brain sugar exerts a regulatory effect too, it means that the central mechanism is not only turned on to combat hypoglycemic emergency, it is all-time active although not in the all-out mode. Unfortunately, there is little research in this direction to confirm the reproducibility of these results.
12.
Hyperphagia, type 1 thrift: glucose sensing neurons in the brain have an important role in regulation of food intake [99, 100]. In the hypothalamic arcuate nucleus, glucose has excitatory actions on anorexigenic POMC neurons, while the appetite-promoting NPY neurons may be directly inhibited by glucose. If the rate of diffusion from blood to brain is slowed down, the “stop feeding” response would be substantially delayed leading to overeating. This could be a mechanism of thrift, type 1 thrift in particular. Since type 1 thrift could be adaptive for socially subordinate individuals, we may expect that social subordination would reduce BBB glut-1. This has never been tested but is also not very difficult to test in appropriately designed experiments combining social behavior and brain chemistry.
13.
The adaptive importance of ketone bodies: The glut-1 hypothesis also suggests a possible functional role for diabetic ketosis. Ketone bodies are made by the liver in advanced diabetes or during starvation. They are derived from fatty acids. A traditional explanation for ketosis is that when tissues are unable to utilize glucose as a fuel due to starvation, insulin deficiency, or insulin resistance, fatty acids are utilized and ketosis is either an inevitable product of fatty acid metabolism or is produced as alternative fuel for tissues. This cannot be true for many reasons. Most tissues can utilize fatty acids as fuels but do not generate ketone bodies. The enzymes required for ketogenesis are specifically located in liver cell mitochondria [101–103]. This appears to be a specific adaptation to provide an alternative fuel for the brain since the brain is unable to utilize fatty acids as fuel but can use keto acids efficiently. Although other tissues can also utilize keto acids, for the brain, in the absence of glucose, it is the only option available. However, there is a paradox here. If keto acids are crucial for the brain in the absence of glucose, why are they produced when there is excess glucose in the blood? Brain is not dependent on insulin for glucose uptake and therefore should be able to take up glucose as a fuel even in an insulin-resistant state. In diabetes, ketone bodies will be needed by the brain only if it does not get sufficient glucose supply. This can happen either in extreme hypoglycemia or when glucose transport to the brain is impaired. If ketosis takes place when there is high or normal blood sugar, we can infer that glucose transport to the brain must be impaired. It may be a specific adaptive response when glut-1 levels are lowered since it results in reduced glucose transport to brain and fatty acids cannot be utilized by the brain tissue [104]. To the best of my knowledge, there is no other ultimate cause for a ketogenic response to evolve.
A possible additional factor that might drive ketoacidosis is as follows. At extremely low glut-1 levels, any other stress or disturbance that can cause sudden increase in brain glucose consumption or drop in availability triggers the sympathetic response. This increases liver glucose production to compensate for low glucose availability to the brain. However, the rate of gluconeogenesis has a physical limit. Also, since glut-1-mediated transport follows a saturation curve, much higher rise in blood glucose would be needed to bring about a small rise in transport to the brain (Fig. 12.6). Simultaneously ketogenesis is stimulated as an alternative rescue mechanism. Both the responses are mediated by the counterregulation machinery. Ketogenesis is able to give some rescue. If ketogenesis fails to start for any reason, another condition called hyperglycemic, hyperosmolar non-ketotic state (HHNKS) ensues. If this model is correct, we can expect that HHNKS should be more dangerous than ketosis. This indeed appears to be true as shown by a comparative study that had a 5% mortality rate in ketoacidosis and 15% for HHNKS [105]. Also, as required by the glut-1 hypothesis, both the conditions are characterized by intense expression of the components of counterregulatory response [106–109]. The traditional line of thinking does not adequately explain why the sympathetic counterregulatory response is triggered when blood sugar is high.
There is one more reason to believe that the ketosis and HHNKS states are driven by central and not peripheral mechanisms. The extremely high levels of plasma sugar reached in these states cannot be explained by insulin resistance and/or insulin deficiency. This is demonstrated by LIRKO mice which have extreme liver insulin resistance but only moderate hyperglycemia that ultimately returns to normal and also by liver parenchyma cells in culture. The difference between gluconeogenesis of liver cells in vitro in the presence (physiological concentrations) versus absence of insulin is of the order of 20% only [110]. This means that even extreme liver insulin resistance or insulin deficiency cannot give the exceedingly high levels of plasma sugar seen in these conditions. The mechanisms have to be other than IR-RII.
If ketogenesis is adaptive, why does insulin, which suppresses ketogenesis, help in such cases? The answer may lie not in ketogenesis but in the hemodynamic action of insulin. Insulin has both short-term and long-term effects enhancing blood flow to tissues which is reduced in insulin resistance [111–113]. Infusion of extra insulin is likely to be helpful in increasing cerebral blood flow and thereby glucose as well as keto acid transport to the brain. This would restore energy flow to the brain and rescue the ketotic condition.
A very important interpretation of the model above is that the pathological effects correlated with hyperglycemia may not be the effects of hyperglycemia but the effects of the relative neuroglycopenia that is causative to hyperglycemia. A condition like HHNKS does not precipitate due to extreme insulin resistance. It happens because of a counterregulatory response to acute neuroglycopenia, and the neuroglycopenia itself could be responsible for the fatal consequences and not hyperglycemia.
Although the glut-1 hypothesis potentially explains over a dozen observed phenomena unexplained by the traditional theory, so far, glut-1 dynamics has not been demonstrated in humans, and therefore, it can be said to be purely speculative. Yet there is some evidence pointing in this direction. Many elements of this story have been demonstrated, although the complete story is yet to be demonstrated. High plasma insulin, which has been shown to enhance cognitive function, increases the rate of brain glucose utilization [114], and this rise is mostly marked in the cortical areas, whereas in the cerebellum, known to be more important in muscle coordination, it is marginal [114]. This supports the notion that brain glucose demand may be increased specifically for cognitive function in diplomat life marked by hyperinsulinemia. Availability of glucose to the brain via the plasma affected cognitive functions in a number of studies [115–119] supporting our notion of the importance of brain glucose in a cognitive brain-dependent diplomat life.
If maintaining a desired level of brain sugar is the objective of regulating plasma sugar, we would expect the brain sugar to remain constant in diabetic patients, although their blood sugar may vary substantially. This has not been tested extensively, but there are some suggestive data. In a rat study, brain glycogen levels did not change in diabetic rats in spite of high plasma glucose, whereas infusing plasma glucose in normal rats increased brain glycogen. Since brain glucose uptake is insulin independent, this difference is not explained by insulin resistance. The difference in the effect of high plasma sugar in normal and diabetic rats suggests that diabetes makes some difference in glucose transport to the brain so that even with rise in plasma glucose, there is no excess glucose in the brain to increase glycogen synthesis [120]. In another comparative study of Zucker obese and Zucker diabetic fatty rats versus lean rats, there was lower brain glucose metabolism, TCA cycle, and glycogen content in obese and diabetic rats than in lean rats [121]. In human studies, there are no data on glut-1 levels, but the ratio of glucose concentration in various regions of the brain to fasting plasma levels can be expected to be lower in diabetics than healthy controls if the BBB glucose transport is slower in diabetics. This is seen in a study where the mean plasma sugar levels of diabetics and nondiabetics were markedly different (9 and 5 mmol/l, respectively), but the brain concentrations were not significantly different [122].
It is very likely that a number of researchers might have suspected some role of brain glucose and looked at brain glucose levels in diabetes. But they did not find anything wrong there and therefore thought that brain glucose was not important in the pathology of T2D. I suspect many researchers might have given up investigating brain sugar in T2D thinking that it was futile, often keeping their results unpublished. This reminds me of the famous Sherlock Holmes story the “Silver Blaze” and the famous dialogue:
Gregory (Scotland Yard detective): “Is there any other point to which you would wish to draw my attention?”
Holmes: “To the curious incident of the dog in the night-time.”
Gregory: “The dog did nothing in the night-time.”
“That” said Holmes, “was the curious incident.”
If the brain glucose does not change substantially in diabetic hyperglycemia, there are important implications of the dog not barking. Minimal changes in brain glucose in spite of diabetes are a significant clue in itself and can be taken as an evidence that brain glucose is the real target of homeostasis. Perhaps there are many mechanisms that buffer brain glucose dynamics from fluctuations in plasma glucose dynamics [59]. Manipulating plasma glucose and restricting BBB glucose transport are among the possible mechanisms of this buffering effort. It can be calculated from the Heikkilä et al. [122] data that under fasting conditions, the ratio of brain sugar to plasma sugar in different parts of the brain was markedly lower in diabetics than nondiabetics (cerebellum 0.51 and 0.86, cortex 0.29 and 0.42, white matter 0.25 and 0.29, and thalamus 0.25 and 0.36, respectively). In another human study, the authors claim to contradict this finding, but the actual data do not. In this study (n = 14 each), after keeping the blood glucose level constant, glucose levels in the occipital cortex of diabetics were lower than that of controls, but the difference was not significant [123]. Since four out of five uppermost brain glucose concentrations belonged to controls and four out of five lowermost to diabetics in these data, it may be suspected that the failure to see significant difference may be due to small sample size and large individual variation. The authors’ conclusion that diabetes does not affect brain glucose dynamics may therefore be too premature. In another study (7 diabetic 11 control), when plasma glucose was increased by 12 mmol/l, brain glucose rise was less in diabetics as compared to controls in all three regions monitored, namely, cortex (2.7 ± 0.9 mmol/l in control versus 2.0 ± 0.7 mmol/l in diabetics), thalamus (2.3 ± 0.7 mmol/l in control versus 1.1 ± 0.4 mmol/l in diabetics), and white matter (1.7 ± 0.7 mmol/l in control versus 1.3 ± 0.7 mmol/l in diabetics), but only thalamus was significantly different [124]. Although individual comparisons were not statistically significant in these studies, the consistent trend of brain sugar rise in diabetics being less than control warrants more attention. In the other direction, there is evidence that similar to rats, even in humans, hypoglycemia is associated with increased rate of glucose transport to the brain [125–127]. So, although there are no data directly on glut-1 levels in humans, the rate of glucose transport across BBB appears to hold an inverse relationship with plasma glucose levels similar to that in rats.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


