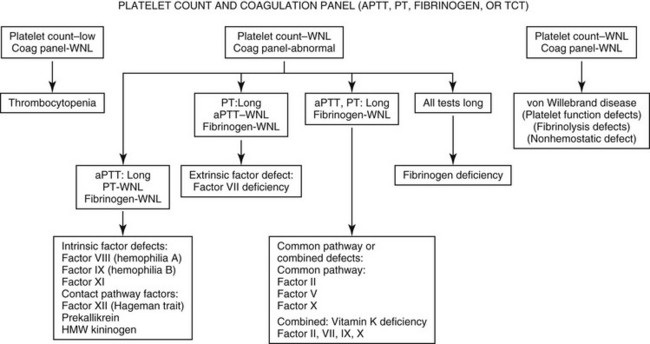
Chapter 62 Clinical signs overlap among the various factor deficiencies, with abnormal gingival hemorrhage at teething, prolonged hemorrhage after surgery or trauma, and recurrent episodes of hemorrhage being common to all (Brooks, 1999). The anatomic site and type of spontaneous hemorrhage may provide some diagnostic clues. Mucosal hemorrhage (e.g., epistaxis, hematuria, melena) is suggestive of VWD, whereas hemarthrosis, intramuscular and subcutaneous hematoma formation, and hemorrhage into body cavities are more typical of coagulation factor deficiencies. Severe forms of VWD and factor deficiencies usually manifest within the first few months of age and patients are transfusion-dependent for survival. Fatalities result from hemorrhage into the central nervous system and respiratory tract or from acute blood loss anemia and hemorrhagic, hypovolemic shock. Milder forms may be unapparent until a patient undergoes surgery or major trauma. The initial workup of any patient suspected of having a bleeding diathesis should include platelet count and coagulation screening tests (Figure 62-1). Consistent use of a standard history questionnaire (Box 62-1) and collection of pretreatment citrate plasma samples facilitate early recognition of a hereditary hemostatic defect and provide the appropriate sample type for definitive testing. For patients with a normal platelet count, abnormal clotting times in one or more coagulation screening tests are compatible with a coagulation factor deficiency. In contrast, dogs and cats with VWD typically have normal coagulation panel results (Johnson, Turrentine, and Kraus, 1988). Buccal mucosal bleeding time is a point-of-care technique to assess von Willebrand factor (VWF) and platelet function. Prolonged bleeding time (> 6 minutes) is a typical feature of VWD. However, the bleeding time end point is also influenced by platelet count, hematocrit, and blood viscosity, and is subject to interoperator variability. The Platelet Function Analyzer (PFA100), a tabletop instrument, has replaced bleeding time determinations as a screening tool to assess primary hemostasis in medical settings. The PFA100 Collagen/ADP closure time is sensitive to VWF deficiency, and prolonged closure times (> 120 seconds) have been described in canine VWD (Mischke and Keidel, 2003). Many of the nonspecific factors that influence bleeding time also affect the closure time end point. Bleeding time and closure times are normal in patients with hereditary coagulation factor deficiencies. Definitive diagnosis of VWD and hereditary coagulation factor deficiencies is based on specific quantitative and/or functional assays of the individual factors. Differences among species in the antigenic and procoagulant properties of hemostatic proteins require species-specific assays or specific validation of human assays for animals. Plasma VWF is primarily measured in immunologic assays that detect protein concentration, referred to as VWF antigen (VWF : Ag). Functional assays have also been developed to measure VWF protein’s ability to bind collagen (VWF collagen-binding activity [VWF : CBA]) (Callan, Giger, and Catalfamo, 2005). Coagulation factors are typically measured in functional clotting time tests that determine individual procoagulant activities (Brooks, 1999). By convention, results of VWF and coagulation factor assays are reported as a percentage of “normal.” Results of each test sample are compared with a plasma standard that has an assigned value of 100%. Values below 50% of the standard indicate a factor deficiency. Canine and feline factor assays are routinely performed in the author’s laboratory for clinical diagnosis of VWD and coagulation factor deficiencies (Comparative Coagulation Section, Cornell University). Information on the biologic basis and inheritance of these disorders is presented in the following sections and in Tables 62-1 and 62-2. TABLE 62-1 von Willebrand Disease Subtype Classification TABLE 62-2 Inheritance Pattern and Coagulation Screening Test Abnormalities for Hereditary Factor Deficiencies The bleeding tendency of VWD is caused by quantitative and functional deficiencies of VWF, a large, multimeric plasma glycoprotein that supports platelet adhesion and aggregation at sites of vascular injury under conditions of high shear (Johnson, Turrentine, and Kraus, 1988). Endothelial cells are the primary source of plasma VWF. In some species, platelets contain a secondary pool of circulating VWF. This pool is lacking in dogs. The highest-molecular-weight forms of VWF, consisting of more than 100 VWF subunits, are the most hemostatically active. Plasma VWF is also a carrier protein for coagulation factor VIII (FVIII), and the association between these two proteins may help localize coagulation to sites of platelet aggregate formation. In another species difference, canine FVIII appears more stable in the absence of VWF than does human FVIII. A subtype classification scheme for VWD in people is applicable for characterizing VWD in animals (see Table 62-1). The classification criteria include VWF concentration, multimeric structure, and interactions with platelets, collagen, and FVIII. Although VWD is the most common hereditary bleeding disorder in dogs, only a few cases have been identified in cats. Type 1 VWD is a quantitative protein deficiency found in many individual canine breeds across many breed groups (see Table 62-1). The clinical expression of type 1 VWD varies and is generally related to the severity of protein deficiency. Dogs with low residual protein concentration (e.g., VWF : Ag < 25%) have the most risk of abnormal bleeding. Additional undefined factors influence clinical expression of this subtype, even among dogs with VWF : Ag below 10%. Types 2 and 3 VWD are more severe forms. Affected dogs often experience spontaneous hemorrhage and are at risk for severe, fatal hemorrhage if they undergo surgery without transfusion. To date, type 2 VWD has been identified only in German shorthaired and wirehaired pointers. Affected dogs have low VWF concentration and the residual protein lacks high-molecular-weight multimers. Dogs with type 3 VWD have essentially no detectable plasma VWF (VWF : Ag < 0.1%). VWD is an autosomal trait. In this form of inheritance, males and females express and transmit the defect with equal frequency. Types 2 and 3 VWD demonstrate recessive expression patterns; homozygous dogs have a bleeding tendency, whereas heterozygous carriers are clinically normal. Type 1 VWD in people is usually a dominant, incompletely penetrant trait. Patients are typically heterozygous or compound heterozygous for VWF mutations. The variable expression of type 1 VWD in dogs is suggestive of this form of inheritance. Regardless of VWD subtype, dogs with VWF deficiency (i.e., VWF : Ag < 50%) can be considered carriers of the VWD trait; to reduce prevalence of VWD within a breed or line, these dogs can be used selectively or removed from breeding programs. However, the VWF : Ag values of carriers may overlap with clear dogs at the low end of normal range. Distinct VWF mutations have been described in Scottish terriers (Venta, Li, and Yuzbasiyan-Gurkan, 2000) and Kooiker dogs (van Oost, Versteeg, and Slappendel, 2004) with type 3 VWD. Direct DNA tests are commercially available for these mutations and for unreported mutations in other breed-variants of VWD (VetGen, http://www.vetgen.com/). Using these tests, dogs with a single copy of a VWF mutation are considered carriers, and dogs with two copies are considered affected with VWD.
von Willebrand Disease and Hereditary Coagulation Factor Deficiencies
Clinical Signs
Diagnostic Strategy
Type
VWF Defect
Affected Breeds
1
Partial quantitative deficiency; residual VWF has normal structure & function
Airedale, Akita, Bernese mountain dog, dachshund, Doberman pinscher, German shepherd, golden retriever, greyhound, Kerry blue terrier, Manchester terrier, miniature pinscher, papillon, Pembroke Welsh corgi, poodle, and others
2A
Selective loss of large VWF multimers, decreased platelet-VWF & collagen interactions
German shorthaired and wirehaired pointers
2B
Increased VWF-platelet binding
Not identified
2M
Decreased VWF-platelet binding with normal VWF multimers
Not identified
2N
Decreased VWF-factor VIII binding
Not identified
3
Complete VWF deficiency
Kooiker, Scottish terrier, Shetland sheepdog
Sporadic cases: Border collie, Chesapeake Bay retriever, cocker spaniel, Labrador retriever, Maltese, Pomeranian
Defect
Inheritance Pattern
Screening Test Prolongation
Hypofibrinogenemia, dysfibrinogenemia
Autosomal
ACT, aPTT, PT, TCT
Factor II (prothrombin deficiency)
Autosomal
ACT, aPTT, PT
*Factor V deficiency
Autosomal
ACT, aPTT, PT
Factor VII deficiency
Autosomal recessive
PT
Factor VIII deficiency (hemophilia A)
X-linked recessive
ACT, aPTT
Factor IX deficiency (hemophilia B)
X-linked recessive
ACT, aPTT
Factor X deficiency
Autosomal recessive
ACT, aPTT, PT
Factor XI deficiency
Autosomal recessive
ACT, aPTT
Factor XII deficiency (Hageman trait)
Autosomal recessive
ACT, aPTT
Specific Disorders
von Willebrand Disease
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


