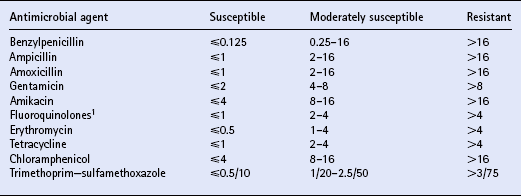
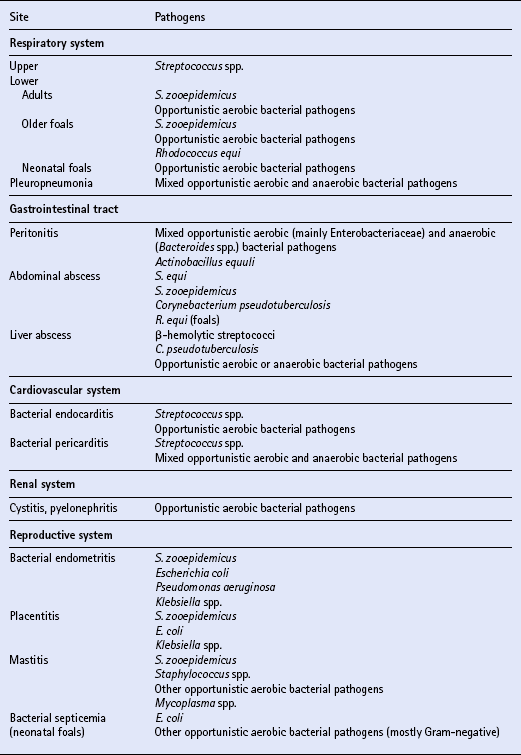
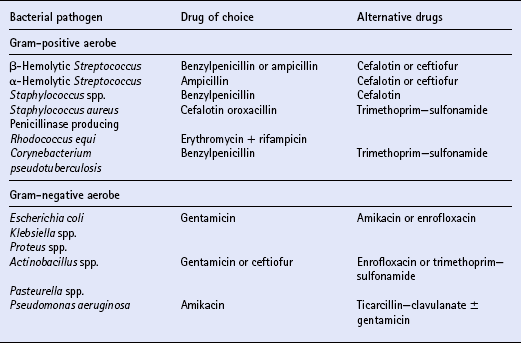
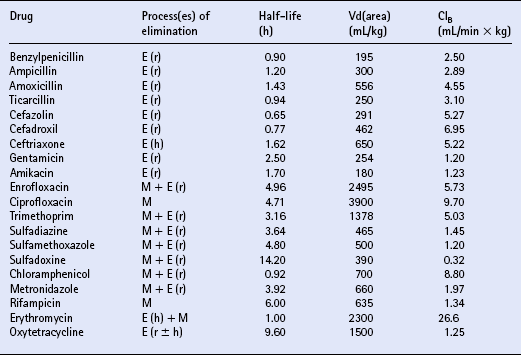
Chapter 4 T.R. Ayliffe, J.D. Baggot, F.M. Cunningham, E. Houghton, Q.A. McKellar, A.M. Nolan, J. Price and R.B. Williams ANTIMICROBIAL DRUG SELECTION AND DOSAGE Quantitative bacterial susceptibility Drug selection for empirical therapy Interpretation of bacterial susceptibility Bioavailability, distribution and elimination NON-STEROIDAL ANTI-INFLAMMATORY DRUGS Anti-inflammatory drug classification Pharmacologic actions of NSAIDs Side effects and toxicity of NSAIDs Pharmacokinetics and administration of NSAIDs Pharmacokinetic—pharmacodynamic modelling of NSAIDs General properties of corticosteroids Anti-inflammatory actions of glucocorticoids Pharmacokinetics and administration DRUGS AND THE COMPETITION HORSE The use of horses in competitions also poses problems, as the several authorities which control equestrian sports each have their own regulations regarding the presence of drugs that could potentially affect the performance of the competitors. Veterinarians must be aware of the regulations and of those drug disposition properties that permit effective use without contravening guidelines. The laboratory results of bacterial culture and MIC determination (when required) give precise data that may be used in selecting the antimicrobial drug, its route of administration and dosing rate for continuation of treatment (specific therapy). The usual dosage regimen for an antimicrobial product is based on providing plasma concentrations of the drug that will be effective against the majority of susceptible microorganisms and will not cause adverse effects in the horse. The assumption is made that the systemic clearance of the drug is not changed by the disease state, even though this may not always be the case. When selecting the antimicrobial drug with which to commence therapy, knowledge of the pathogenic bacteria that most commonly cause infections at various locations is useful but should be supported by examination of a stained (Gram or Romanowsky-type Wright–Giemsa) direct smear. Bacterial pathogens that have been isolated from various sites of infection are listed in Table 4.1. Because the principal bacterial species isolated from any site of infection may vary with geographic region, specimen collection for bacterial culture prior to commencing antimicrobial therapy is of paramount importance. The suggested drug of choice and alternative drug for initial treatment of infections caused by various microorganisms are presented in Table 4.2. The information in Table 4.2 should be used in conjunction with direct smear examination for drug selection (empirical therapy) while awaiting the results of bacterial culture and quantitative susceptibility testing (when considered necessary). Selection of the drug to use is influenced by location of the site of infection. There are no agreed guidelines for the interpretation of quantitative susceptibility results for equine bacterial pathogens. Suggested interpretative guidelines for MIC breakpoint values are presented ( Table 4.3). The categories are: Table 4.3 1Quantitative susceptibility of bacterial pathogens varies between individual fluoroquinolones. 1. Susceptible, in which an infecting organism is inhibited by concentrations achieved in blood or tissue fluid after the recommended dose including oral administration 2. Moderately susceptible, when an infecting microorganism is inhibited only by blood concentrations achieved with maximum doses, up to the limit of toxicity 3. Resistant, when an organism is resistant to usually achievable concentrations of the antimicrobial drug in blood or tissue fluid. The MIC (q.v.) is the quantitative value normally used to define the in vitro susceptibility of a microorganism. It is used, in conjunction with pharmacokinetic parameters that describe the bioavailability and disposition of antimicrobial agents, to calculate appropriate dosing rates. This approach allows appropriate dosage adjustment to be made for organisms of different susceptibilities. It is the basis for calculating optimal dosage. Even though quantitative susceptibility (an in vitro measurement of activity) generally correlates with clinical efficacy, it cannot be guaranteed to predict the response to therapy. Furthermore, use of the antimicrobial agent to which a pathogenic microorganism is most susceptible in vitro might not be clinically indicated because of its potential to produce adverse effects. The action of some antimicrobial agents, whether administered orally or parenterally, on the cecal and colonic commensal microorganisms of horses can cause serious digestive disturbances. This adverse effect makes penicillins unsuitable for oral administration to horses other than newborn foals and limits the use of macrolides and, under certain circumstances, of tetracyclines. Foals appear to be less susceptible than adult horses to disturbance in the balance of commensal microbial flora in the large intestine. It is likely that the microbial balance is related to the composition of the usual diet. When the rate of absorption significantly influences the overall rate of elimination of a drug, flip-flop pharmacokinetics apply, allowing the use of a longer dosage interval than that associated with a conventional (immediate release) preparation of the drug. This applies to procaine benzylpenicillin, which is an aqueous suspension. Following the administration of a single dose (20000IU/kg) of procaine benzylpenicillin at various IM injection sites and SC, the peak plasma concentration (Cmax) and systemic availability (indicated by area under the curve over a 24 h period) of benzylpenicillin were highest when the long-acting preparation was injected IM in the neck region (m. serratus ventralis cervicis) and were lowest when it was injected SC. Having traversed the gastrointestinal mucosal barrier, drug molecules are conveyed in hepatic portal blood to the liver, where they are subjected to the “first-pass” effect prior to entering the systemic circulation. Hepatic first-pass metabolism can substantially decrease the systemic availability of lipid-soluble drugs that undergo microsomal oxidative reactions (e.g. trimethoprim, metronidazole, rifampicin, enrofloxacin). While the metabolites of most antimicrobial agents are inactive, some, such as desacetylrifampicin and ciprofloxacin (to which enrofloxacin is converted), have antimicrobial activity at least equal to that of the parent drug. Because the formation of desacetylrifampicin is capacity-limited, the half-life of rifampicin is dose-related in horses. Certain antimicrobial agents (erythromycin and chloramphenicol) inhibit hepatic microsomal enzyme activity, whereas rifampicin is a potent inducer of hepatic microsomal enzymes. Drug-induced changes in microsomal enzyme activity are generally associated with chronic rather than short-term use of activity-modifying drugs. Amoxicillin distributes more widely than benzylpenicillin in extravascular fluids and tissues, presumably due to the higher lipid solubility of amoxicillin. Penicillins, with the exception of nafcillin, are eliminated by renal excretion (glomerular filtration and proximal tubular secretion). Nafcillin and a small fraction of amoxicillin in the systemic circulation are excreted in bile. The β-lactamase inhibitors (clavulanic acid and sulbactam) do not alter disposition of the penicillins with which they are combined in commercially available dosage forms. β-Lactamase inhibitors enhance the activity of aminobenzylpenicillins (amoxicillin, ampicillin) and ticarcillin. The overall elimination of antimicrobial agents obeys first-order kinetics when usual doses are administered by IV injection. This implies that 50% of the drug in the body is eliminated (by metabolism and/or excretion) each half-life. Because lipophilic antimicrobial agents are eliminated mainly by the liver (metabolism or biliary excretion) their half-lives vary widely compared with the half-lives of drugs that are eliminated by renal excretion ( Table 4.4). Even though oxytetracycline is eliminated by renal excretion, the relatively long half-life of this antibiotic is attributed to enterohepatic circulation. An application of half-life is in the selection of an appropriate dosage interval, which may be either approximately equal to or a small multiple of the half-life. This is especially important for antimicrobial agents that produce a bacteriostatic effect (e.g. tetracyclines, chloramphenicol, sulfonamides).
Toxicology and pharmacology
INTRODUCTION
ANTIMICROBIAL DRUG SELECTION AND DOSAGE
MICROBIOLOGIC CONSIDERATIONS
Approach to therapy
Location of infection
Drug selection for empirical therapy
Interpretation of bacterial susceptibility
PHARMACOLOGIC CONSIDERATIONS
Routes of administration
BIOAVAILABILITY, DISTRIBUTION AND ELIMINATION
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Toxicology and pharmacology
Only gold members can continue reading. Log In or Register to continue
