(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
If we are set to search for an alternative picture, where do we need to begin our search? There is a simple and universal answer to all such questions in biology. Whenever any seemingly paradoxical and intuitively difficult phenomenon is observed in biology, we need to ask why and how it might have originated in evolution. Evolution is fundamental to biology, and as rightly put by the evolutionary geneticist Dobzinski, “Nothing in biology makes sense except in the light of evolution.”
Introduction
If we are set to search for an alternative picture, where do we need to begin our search? There is a simple and universal answer to all such questions in biology. Whenever any seemingly paradoxical and intuitively difficult phenomenon is observed in biology, we need to ask why and how it might have originated in evolution. Evolution is fundamental to biology, and as rightly put by the evolutionary geneticist Dobzinski, “Nothing in biology makes sense except in the light of evolution.”
But there is a great danger lingering around. Many people frequently use a trivial version of evolutionary logic to somehow “explain away” things. There is no dearth of “just so…” evolutionary stories popular around including the loss of tail, predicted loss of nails and eyebrows in humans, the evolutionary fate of today’s monkeys, the long neck of giraffe, and so on. Therefore one needs to be rigorous while trying to apply an evolutionary explanation to anything. Fortunately mainstream evolutionary biology has matured sufficiently today and has developed methods that can save us from falling into such traps.
If we are going to look for an evolutionary theory for T2D and related disorders, let us first list our expectations from an evolutionary theory. (1) It should address the basic riddle why a disorder that has strong familial tendencies and so has some presumably genetic components has suddenly increased its prevalence in the last couple of generations. (2) It should explain the apparent polymorphism in the population, i.e., individual differences in the tendency to become obese and/or diabetic. (3) It should explain the observed epidemiological associations and patterns, for example, the strong association between birth weight and metabolic syndrome. (4) It should not stop at explaining the origins of obesity but also explain why obesity is associated with insulin resistance and its consequences with both proximate and ultimate components of reasoning; more specifically, there needs to be an ultimate reasoning for why obesity induces insulin resistance (if it does) and other metabolic and endocrine changes. (5) It should give logical and testable solutions to all the paradoxes that we saw in the last chapter. (6) It should help us find out why T2D is found to be largely “incurable,” at least with the current therapeutic approach. (7) It should be falsifiable and either be supported by existing evidence and/or make a series of predictions that can be tested sooner or later. (8) Optimistically (but optionally), it should lead to some fundamental breakthrough of clinical importance. If it leads to a long-term solution or “cure” for diabetes, sooner or later, it would be really fascinating. This cannot be the primary objective of an evolutionary theory though. Evolutionary theory is not an application-oriented science. Its main objective is to develop fundamental insights into biological reasoning. However, as our insights increase, it is very likely that we find an application.
Origin of Thriftt
Evolutionary approaches to obesity and T2D are not new. There is a history of evolutionary hypotheses intended to do one or more of the above. The first major change in the thinking about diabetes that had a strong evolutionary flavor was brought about single-handedly by a person called James Neel in the early 1960s [1]. Most people at that time believed that diabetes was a genetic disorder much like other well-known genetic disorders such as sickle cell anemia or hemophilia. The belief was so deep rooted that there are papers claiming Mendelian nature of inheritance of type 2 diabetes [2–4], although we know now that T2D is far from being Mendelian. This amusing conclusion appears to arise out of some facets of human nature to which researchers are no exception. When there is a theory and you expect to see or find something based on it, frequently, you start seeing things although they may not actually be there. Also if you do not expect something or do not have a theory for it, you are very likely to miss seeing it, although it may very much be there (“We do not see what we sense. We see what we think we sense” [5]). I have experienced this frequently with undergraduate students. When they are told that the given sample is most likely to contain say staphylococci, every dust particle starts looking like Staphylococcus. Perhaps this is how the Mendelian inheritance of diabetes was “observed” by some researchers. Neel also appears to believe quite strongly that inheritance of diabetes was Mendelian. Still Neel appears to be the first person to be convinced that diabetes could not be a “genetic disorder.” Not that he doubted the “genetic” part of it, but he raised doubts about diabetes being a “disorder” of genetic origin. All monogenic (where a defective allele at a single gene locus can be clearly shown to be responsible for the disorder) heritable disorders always exist in any population in exceedingly small frequencies, typically one in several thousand. Diabetes, on the other hand, was much more common and notably increasing in frequency by this time. Realizing that no genetic disorder can be as frequent as diabetes is and moreover cannot increase in a population so rapidly, he thought that diabetes should not be treated as a genetic disorder. If the frequency of the diabetic allele is high, there must be a biological reason for it. A frequency of a gene can increase if it is useful in some way so that natural selection favors individuals having that allele. Therefore Neel thought that the genes responsible for diabetes must be adaptive under at least some set of conditions. Since the association of obesity with diabetes was well accepted, he equated the diabetic tendency to the tendency to accumulate fat and argued that a “thrifty” gene helped storage of fat under conditions of better availability of nutrients and allowed reutilization of it under starvation. Neel’s definition of thriftiness is “being exceptionally efficient in intake and/or utilization of food.” This gives two distinct meanings to thrift. Either a thrifty individual eats more and stores fat or has a less wasteful metabolism which allows greater storage of energy. Let us call the intake efficiency as type 1 thrift and utilization efficiency, i.e., lower rate of burning calories to store more, as type 2 thrift. This classification is extremely important to understand thrift, but most researchers have failed to clearly differentiate between the two, leading to much ambiguity. The two are mutually compatible, and Neel appears to imply both. But as we will see below, both do not apply equally to all arguments about thrift. Therefore, for every argument, there needs to be some clarity as to whether we are talking about type 1 or 2 thrift or both. In Neel’s view the thrifty gene was under positive selection pressure in ancestral life when seasonal and climatic conditions resulted into fluctuating food availability often called “feast and famine.” The assumption is that human ancestors frequently faced periods of starvation interspersed with periods of food abundance. A genetic tendency to store fat when food was abundant was extremely adaptive under feast and famine conditions and therefore enjoyed a selective advantage. In modern life food is always abundant, something that can be called a “feast and feast” condition, and under such conditions, the same genetically determined tendency leads to obesity. Both types 1 and 2 thrift are compatible with this argument. According to this theory, obesity is the apparent cause of diabetes, but the diabetic tendency is the real cause of obesity. Neel appears to have believed that diabetic tendency leads to obesity. In his own words, “The overweight individual of 40 or 50 with mild diabetes is not so much diabetic because he is obese, as he is obese because he is of a particular (diabetic) genotype.” What Neel refers as mild diabetes is type 2 diabetes in the current nomenclature. The parenthesis for the word “diabetic” is original. Neel’s argument was based on a number of patterns well known by then including that diabetic mothers (also diabetic fathers!!) have larger babies, that hyperinsulinemia precedes adult-onset diabetes, that insulin has lipogenic action, and also that frequently there is an early history of transient hypoglycemia much before diabetes sets in. Neel thought that hyperinsulinemia is the thrifty tendency. High levels of insulin induce lipogenesis and fat accumulation since it facilitates entry of glucose into fat cells and its conversion to fat. One of his relied evidence was an early hypoglycemic phase in the history of T2D. This hypoglycemia perhaps represented active lipogenic metabolism under the influence of insulin. He thought that at later stages, the high levels of insulin were compensated by “anti-insulin” activity (which could be insulin resistance in today’s terms), and eventually, somehow, anti-insulin activity became stronger than insulin action to cause diabetes.
Although I am going to argue against some of Neel’s concepts, I must admit first that this paper is one of the greatest papers ever written on diabetes so far. It is full of prophecy and foresight. Neel does not hesitate to make bold speculations whenever he is confident about the logic behind them. It is doubtful whether in today’s science publishing trend, a paper as speculative as this one would have ever got published. Speculations are generally unwelcome in today’s science, and speculative papers are most likely to be rejected by the current editorial standards. Perhaps things were different in the early 1960s or Neel was extremely lucky. Neel’s speculations had a far-reaching vision. Interestingly when Neel wrote this paper, the concept of insulin resistance was not yet established. But Neel appears to have perceived it speculatively and calls it “anti-insulin” activity. Neel also had a very clean insight into a number of issues with which the followers of the thrifty gene hypothesis later messed up completely. Neel seems to be clear in his mind that hyperinsulinemia comes first and anti-insulin activity appears consequently. He also has rejected the idea of β cell “exhaustion” leading to diabetes.
Neel’s concept of thrift soon became almost an axiom, although no such “thrifty” gene or set of genes have been convincingly demonstrated any time. Later the observation that individuals born small for gestational age had a greater probability to become obese and type 2 diabetic in later life led to the concept of fetal programming [6–10]. This hypothesis states that if a fetus faces inadequate nutrition in intrauterine life, the body is programmed to be “thrifty” as an adaptation. Although a little later I am going to contest this logic as well, let us first appreciate the great vision of the researchers who detected this pattern for the first time. It is not easy even to imagine that the roots of a disorder that becomes obvious in one’s 40s and 50s could have its origins in intrauterine life. But a group of British visionaries discerned such a strange relationship with insightful analysis of data. The concept originated in data on differential death rates in different parts of Britain. Death rates in the newborn in the early decades of 1900s were highest in some of the northern industrial towns and poorer rural areas in the north and west. This geographical pattern closely resembled that in deaths due to coronary heart disease decades later. This led to the suspicion that intrauterine growth retardation had something to do with heart disease in adults. Epidemiological studies were launched based on the simple strategy of examining men and women in middle and late life whose body measurements at birth were recorded. Sixteen thousand men and women born in Hertfordshire during 1911–1930 were traced, and the effort revealed that death rate due to coronary heart disease in the lowest birth weight class was double than in the uppermost class [8]. Later, similar trends were found across most parts of the world and different ethnic groups, showing the robustness of the relationship between fetal growth conditions and adulthood disorders including obesity, type 2 diabetes, hypertension, and coronary heart disease [11–17]. There are two possible components of the proposed thrifty adaptation in response to reduced fetal growth. One relates to an immediate gain in terms of survival during fetal and early infant life. The other is a predictive adaptive response in anticipation of starvation in later life [18]. This distinction is important in understanding the evolution of fetal programming as we will see soon.
Both the concepts of thrifty gene and thrifty phenotype by fetal programming have recently faced serious criticism on several grounds [19–24]. Some of the critics think that the thriftiness concept is flawed and needs to be abandoned [20, 21]. Others have attempted to refine the concept of thrift so as to resolve some of the flaws and paradoxes pointed out by the critics [25–28]. Let us see the classical concept of thrift and its criticism first before considering the refined versions of thrift. Baig et al. [29] integrated the critical arguments challenging the classical thrifty gene and thrifty fetal programming hypotheses using a mathematical model. The model considered three hypothetical genotypes, namely, a non-thrifty wild type having no mechanism for thriftiness, a thrifty genotype which is genetically programmed for thriftiness, and a genotype with a capacity for fetal programming for thriftiness. Taking a year as a natural time unit of seasonality, a simple dichotomy of years was assumed in the model, those with adequate food supply (feast) and those with inadequate food supply (famine). Famines were assumed to occur randomly with some probability. The fitness of an individual with non-thrifty genotype in feast conditions was assumed to be greater than that of an individual with thrifty genotype because of a cost associated with thrift. Obesity and insulin resistance are known to be associated with reduced fecundity [30–33] justifying the cost of thriftiness in feast conditions. Fitness of an individual with non-thrifty genotype in famine conditions was assumed to be less than that of individuals with thrifty genotype. It is simple to visualize from this set of assumptions that at low probability of famine, the non-thrifty genotype will have an advantage, and at high frequency, the thrifty one will have an advantage. It also follows that at no condition, non-thrifty and thrifty genotype would coexist in a stable polymorphism. This is simple to see intuitively as well as to show with simple mathematics as done by the Baig et al. model [29].
Calculation of the fitness of the thrifty programmer was a little more complex. Assuming no correlation between birth and lifetime conditions, the total fitness was written as the sum of all years with the assumption that in the birth year the phenotype was best suited for given conditions. For the rest of the lifetime, the fitness fluctuated according to randomly fluctuating environmental conditions. Analytical solutions to the model showed that at low probabilities of famine, a non-thrifty gene has a selective advantage, and at high probability, thrifty gene would get selected (Fig. 4.1) leaving a very narrow area of advantage for fetal programming. The area was narrower for long-lived species, whereas for short-lived ones, the birth year advantage was large enough as compared to lifetime, and therefore, fetal programming had a much larger width of advantage (Fig. 4.2). Two interesting conclusions of the model are that a thrifty gene can evolve but cannot have a stable polymorphic state and that fetal programming for thrift is most unlikely to evolve by feast and famine conditions in long-lived species [29]. There are a number of other issues about the concept of thrift that need to be addressed.
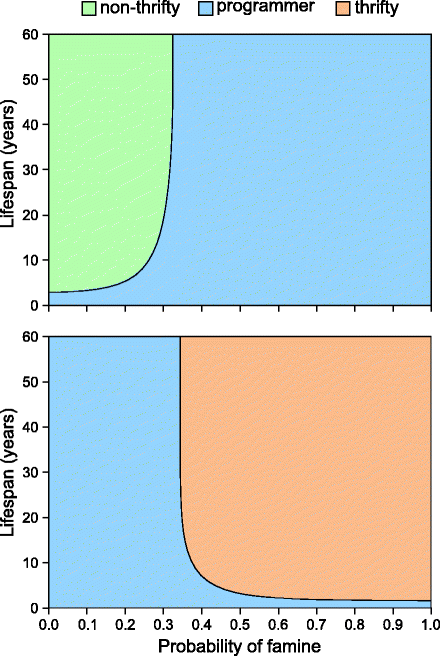
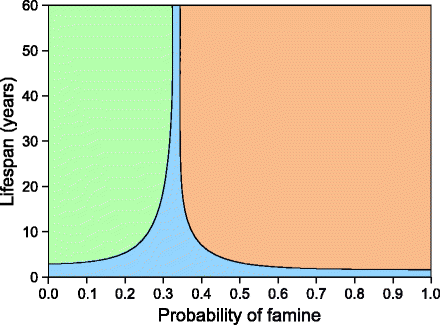
Fig. 4.1
Parameter areas of advantage in the Baig et al. model [29] (upper) when only non-thrifty genotype and fetal programmer compete. (lower) When only thrifty genotype and fetal programmer compete. For this and all other figures in this chapter, the colors denoting advantage areas for the thrifty gene, non-thrifty gene, and fetal programmer remain the same
Fig. 4.2
Parameter areas of advantage when non-thrifty genotype, thrifty genotype, and fetal programmer compete simultaneously: Fetal programming can evolve for species with short life span. If the life span is longer, fetal programming is unlikely to offer selective advantage over thrifty or non-thrifty genotypes except for a specific and very narrow range of probability of famine
1.
Feast and famine conditions in human ancestry: Is the assumption realistic?
The fundamental assumption of the thrifty gene hypothesis is that human ancestors suffered from wide fluctuations in food availability either as an effect of season or of year-to-year climatic fluctuations. Thrift evolved in response to these fluctuations. One of the key questions is when in human history could selection for thriftiness, if any, have operated. There are three possible scenarios:
(a)
Selection during hunting-gathering stage: Contrary to the commonly held belief, paleoarcheological as well as anthropological data suggest that chronic starvation was uncommon during hunter-gatherer stage [34, 35]. Today’s hunter-gatherer societies do not seem to suffer starvation more frequently or more intensively than agricultural societies [22]. This is in spite of the fact that today, hunter-gatherer societies are pushed to marginal or difficult habitats, and most pristine habitats are occupied by modern man. Therefore the assumption that hunter-gatherer societies suffered frequent starvation is not well supported. But even if we assume hunter-gatherer societies to be prone to feast and famine selection, a number of other questions remain unanswered. Since hominids were hunter-gatherers for the most part of human evolutionary history, selection would have been prolonged, and we would expect alleles to have reached equilibrium frequencies. The Baig et al. model [29] implies that selection cannot result in stable polymorphism of thrifty alleles. In the modern human society, there is considerable variation in the tendency to become obese or diabetic. Therefore polymorphism with respect to genes predisposing to obesity and type 2 diabetes presumably exists. If there is no negative frequency dependence or heterozygote advantage, natural selection will be directional resulting into the fixation of the advantageous genotype. At low frequencies of famines, the non-thrifty gene would be the only survivor, and at high frequencies, the thrifty gene would be the only survivor. A critical frequency of famines separates the two, and in no case, the thrifty and non-thrifty genes can coexist stably. Theoretically if a heterozygote of thrifty and non-thrifty alleles gets a dual advantage by expressing the right allele in the right environment, the two alleles can coexist and the population at any given time will consist of thrifty, non-thrifty, and all-time fit heterozygous individuals. Neel assumed that the thrifty gene will coexist in a stable state owing to heterozygote advantage [1]. However, there is no evidence of any heterozygote advantage so far. Stable polymorphism is also possible if there is negative frequency-dependent selection. However if fitness is decided by climatic conditions as assumed by the popular version of thrifty gene hypothesis, frequency dependence is unlikely. Therefore selection during hunter-gatherer stage does not explain the prevalent polymorphism in predisposition to obesity.
(b)
Selection after the beginning of agriculture: Chronic starvation due to famines became more serious and common with the beginning of agriculture [36–38]. Signs of chronic starvation such as linear enamel hypoplasia of teeth are more common in early agricultural societies than in hunter-gatherer societies. This is owing to the fact that crops are highly seasonal in nature, and the failure of a single crop leads to long-term food scarcity. Such long-term food shortages are much less probable in hunter-gatherer life, particularly in biodiversity-rich areas. Therefore if selection for thriftiness started acting after the beginning of agriculture, there could be transient polymorphism. A testable prediction of the hypothesis would be that ethnic groups that took to agriculture earlier should show higher tendencies to become obese and diabetic. Data on ethnic groups such as the Australian Aborigines who remained hunter-gatherers until recently is not in support. The recently urbanized individuals of this community developed a surprisingly high prevalence of diabetes and hypertension [39] indicating that selection during agriculture life is unlikely to be responsible for the spread of a thrifty gene. It is difficult to argue therefore that thrifty genes evolved after the advent of agriculture.
(c)
Selection in modern times: Intensive agricultural and industrial societies are modern phenomenon just over 200 years old, and it is highly unlikely that this period could have brought about any evolutionary change, although this has been claimed (see below). It can be seen from all the three possible scenarios that natural selection for the hypothetical thrifty gene(s) is unable to explain the apparent polymorphism or high variation in obesity proneness.
2.
Does the tendency to become obese protect against a famine?
Whether obese people have a significantly better chance of surviving famines is debatable, but if we assume so, we can use the model to estimate how much advantage is needed for the thrifty gene to evolve. What could be the threshold probability of famine that would permit the evolution of thrifty genotype? Looking at long-term history, Speakman argued that famines with significant mortality occur with a frequency of once in 100–150 years [21]. At this frequency the advantage of thrifty over non-thrifty phenotype in famine conditions should be more than 100 times the relative loss suffered by thrifty gene in feast conditions. Since an obesity-induced reduction in fecundity has been demonstrated, the advantage of thriftiness in famines should be exceedingly large for thrifty genes to evolve. Such a large advantage should be highly evident and easily measurable, but obese people have not been shown to survive famines significantly better than lean individuals [21]. Therefore it is doubtful whether obesity actually offered sufficient advantage during famines to get selected. This is particularly tricky because a major cost associated with obesity is reduced fecundity [30–33, 40, 41]. If there is any advantage of obesity, it should be greater than its reproductive cost, and there have been no attempts to test this quantitatively.
Which particular type of thrift is more relevant here? In principle, both types 1 and 2 thrift may help in a feast and famine or energy limitation problem. But what does evidence show? Which type of thrift is evident in the current obesity epidemic? Although lower metabolic rates have been assumed to lead to obesity, actual measurements of basal metabolic rates have given contradictory results [42–45]. Impaired fat oxidation rather than lower metabolic rate appears to be the main contributor to obesity, and demonstration of impaired fat oxidation in obesity is more consistent across studies [46–50] than basal metabolic rates. Impaired fat oxidation is neither directly implied by type 1 nor by type 2 thrift. If inability to reutilize stored fat is the major cause of obesity, the stored fat is unlikely to help under “famine” conditions, and if this is so, it is a major blow to the thriftiness hypotheses irrespective of whether it is type 1 or 2 thrift. The doubly labeled water studies also suggest that obesity is more a product of hyperphagia than metabolic frugality [51, 52]. Therefore if thrift really exists, it could be type 1. Type 2 thrift stands on very slippery grounds since evidence for lower metabolic rates mainly leading to obesity is laden with contradictions. The known genetic mechanisms of obesity also work by interfering with appetite control rather than through metabolic thrift [53]. Therefore, lower rate of metabolism is unlikely to be the cause of obesity although very widely believed so and the evidence for impaired fat oxidation in obese individuals casts doubt on its usefulness in famine conditions.
3.
Are we really adapted to feast and famine?
A number of animals experience periods of extreme feast and famine in their natural environments. There are two major types among these. One consists of animals that take a summer sleep (aestivation) or winter sleep (hibernation). This happens when the winters or summers are so harsh that foraging becomes impossible. The other situation is long-distance migration. Many long-distance migrants including migratory birds travel thousands of kilometers at a stretch which takes several days during which food intake is either absent or marginal but energy expenditure is large. Before either migration or hibernation, animals store large amounts of fat and utilize them during the nonfeeding period with or without active energy expenditure. These animals have fine-tuned their entire metabolism to suit both accumulation of fat as well as utilization of fat. If humans were adapted to periods of food abundance interspersed with periods of starvation, a set of similar metabolic adjustments should be seen in us too.
When migratory birds utilize their fat, they do so with priority, and they can keep on flying until their entire fat is exhausted. Often when migrating birds are found dead, it is due to complete exhaustion of stored energy. When a fat human performs exercises of any intensity, he feels exhausted soon when hardly any portion of the total fat is burnt. If humans starve to death, a substantial amount of fat remains unutilized demonstrating that we have not evolved to be efficient fat utilizers. Most of the deaths during famines are due to infection rather than due to complete energy exhaustion [20, 21]. Humans are equally inefficient in accumulating fat as compared to migrating birds or hibernating animals [54]. When humans starve, the muscle protein starts breaking down much before fat stores are exhausted, whereas in animals adapted to long-time starvation, muscle proteins are conserved until fat depots are exhausted. Also after refeeding muscle strength is regained first in these animals, and fat starts building up later, whereas as in humans, refeeding after fasting starts building fats rapidly keeping muscles weak. While feast and famine animals can migrate or sleep for several weeks or months without feeding with no symptoms or distress, humans feel hungry and restless only after brief fasting, and several symptoms of starvation start appearing much before any significant depletion of fat stores. All these are certainly not signs of being well adapted to feast and famine conditions [55]. If anything we are the poorest performers among all animals facing feast and famine conditions naturally. It is therefore doubtful whether we have evolved any thrift at all. If we have, it does not appear to be in response to seasonal starvation since we do not show any characteristics of animals adapted to seasonal starvation. Interestingly the species most well adapted to overfeeding and starvation do not show adverse effects of obesity as they accumulate fat every year. This suggests that rather than being thrifty, the failure to be thrifty might be the real cause of our obesity problems.
4.
Is there a genetic tendency to be obese?
A large body of research has now focused on the genetics of obesity. After the human genome sequence, much deeper insights into the genetics of obesity were expected. An increasing number of loci and mutants associated with obesity are being identified from genome-wide association (GWA) studies. However, there are certain internal paradoxes associated with these data. Studies prior to the genomic era that were based on familial, twin pair, and adoption studies typically predicted a large heritable component in obesity [56]. The GWA studies, on the other hand, have identified a large number of associations, but they together explain a very small fraction of variance in obesity parameters [57–64] leaving a large gap between the pregenomic and emerging genomic picture. The pregenomic studies had estimated between 40 and 80% genetic influence on obesity [56]. In contrast although the genome-wide association studies have revealed a large number of potentially important alleles, they together do not explain more than 2–5% of population variance in body weights. Genes that have a stronger influence on a phenotypic character have a greater probability of being discovered early in such studies. Therefore it is highly unlikely that some gene having a large influence on obesity exists but has not been discovered as yet. But one can expect many more loci with small effects yet to be discovered. How may gene loci have alleles that influence BMI or other parameters of obesity? We do not have a definitive answer to this question as yet. Various estimates have been made based on different considerations and different sources of data which range from a couple of dozen to over 6,000 [65]. Although the exact number is not known today, we can say with confidence that a large number of genes are involved in obesity, each with a small effect size.
This itself means that genes cannot be responsible for variability in the population in the tendency to become obese. The logic is so straightforward and robust that it can be stated almost like a theorem. If obesity were to be decided by a single gene with two alleles such that homozygous for the pro-obesity allele was highly obese, homozygote for the antiobesity allele lean, and the heterozygote intermediate, there would be some mean and variance of population obesity distribution. If we make a simplistic assumption that the frequency of the pro-obesity allele (p) and that of antiobesity allele (q) is equal, by binomial distribution, 25% population will be obese, 50% intermediate, and 25% lean. This is not the reality, but we can take this hypothetical case as a baseline for further mathematical arguments. If we give obesity score of zero to the lean, 0.5 to the heterozygote, and 1 to the homozygous obese, the coefficient of variation works out to be unity. Now if we divide the gene effect to n different gene loci each with one pro-obesity and one antiobesity allele and each locus having a magnitude of effect 1/n th that of the original single gene, we will have a more generalized frequency distribution of the population obesity. Using simple mathematics, it can be shown that the coefficient of variation of this distribution will be (√ 2npq/2np). Going by our original assumption of p = q, it simplifies to 1/ √2n. If we go by the most conservative estimate of a couple of dozen gene loci affecting obesity, the coefficient of variation becomes approximately 0.15, indicating that 85% of population variability is lost. If we go by the estimates on the higher side, more than 99% variability is lost. Relaxing the assumption of p = q makes the mathematics more complex, but the conclusion remains the same, i.e., the greater the number of loci, the smaller is the genetic variability in the population.
For readers who do not understand binomial distribution, there is a simple intuitive way to state the same logic. We assume that there are a large number of genes, each with a small effect on obesity and that they segregate independently. Now if I draw a random set of alleles, I will almost invariably get approximately half of them pro-obesity and half antiobesity. It is next to impossible that some individual gets mostly pro-obesity alleles and someone else gets mostly antiobesity alleles. Therefore all individuals will stand at more or less the same level of obesity. The greater the number of genes involved, the greater will be the similarity between individuals. So it can be stated as a theorem that if obesity is a polygenic trait involving a large number of genes with small individual effects, the observed population variation and apparent heritability cannot be genetic.
In short, based on genomic data, it can be safely concluded that genes have a negligible role in the prevalent epidemic of obesity. We are yet to understand the reasons for the discrepancy between the pregenomic and genomic estimates of the genetic component in obesity. Some of the possible answers are epigenetic mechanisms [66, 67], intrauterine effects, transgenerational effects [25], or familial inheritance of dietary and behavioral traits. The failure to detect genetic influence on obesity is a robust evidence against any hypothesis involving a gene or set of genes for thrift. So with GWA, we can now permanently bury thrifty gene hypothesis. This can be interpreted in favor of the thrifty phenotype or fetal programming hypothesis and its variants.
5.
Can lifetime programming for fetal conditions be adaptive?
The thrifty phenotype or fetal programming hypothesis suffers from a different set of problems. Fetal programming can offer two types of potential advantages, short-term survival advantages in fetal and early infant life and long-term predictive adaptive advantages of lifelong duration. If the advantage is of a short duration, it is difficult to explain why a lifetime commitment to a particular metabolic state could have evolved. A number of genes have age-specific expressions. The endocrine and metabolic states of the body change substantially during adolescence, puberty, pregnancy, parenting, menopause, or senescence. This demonstrates the adaptive flexibility of the body with age. Therefore any rigid lifelong programming for short-term advantage is a difficult proposition. If climatic fluctuations were the main selective force, it should evolve metabolic flexibility rather than lifelong rigid programming since climatic conditions may change substantially and unpredictably in one’s lifetime. Although we are assuming that there is a short-term advantage of thrift when faced with fetal or infant undernutrition, such an advantage has never been demonstrated. Are the would-be-diabetic individuals really better adapted for early life malnutrition? So far there is no data demonstrating convincingly any such advantage to the thrifty phenotype.
Metabolic programming of a lifelong duration based on intrauterine conditions is unlikely to offer a fitness advantage except in two sets of conditions. As the Baig et al. [29] model suggests, if a species has a very short life span, fetal programming for adapting to the birth year conditions can be beneficial since the birth year itself is a substantial part of the total life span. Assuming 1 year to be the natural unit of seasonal cycles, species with a life span of <3–5 years can be expected to evolve lifelong fetal programming for thriftiness even though the adaptive advantage is of a short duration. For long-lived species, fetal programming is unlikely to evolve unless there is a significant positive correlation between birth conditions and lifelong conditions (Fig. 4.3). Climatic fluctuations from year to year are a complex phenomenon, and since prediction of important climatic features such as rainfall has important implications, there have been serious attempts to detect temporal patterns. However, temporal patterns are of little help in weather prediction since there are no consistent time-lapse correlations in rainfall or other parameters. Since India has the largest population of diabetics and monsoon is the most important determinant of food availability in this region, it would be enlightening to see the patterns in the Indian monsoon. Table 4.1 shows that rainfall in a given year is not correlated with that of the subsequent year, subsequent 10 years or 40 years cumulative [29]. Over the 30 monsoon subdivisions of India, only 6 are statistically significant using individual α level of 0.05, out of which four correlations are negative contrary to the expectation. Using Bonferroni correction for significance level, applicable when a large number of tests are being done together, none of the correlations remain significant. As there is no detectable positive correlation between birth year and lifetime rainfall conditions and no such correlational patterns in any other climatic variables are reported, fetal programming is unlikely to have evolved in anticipation of drought or famine.
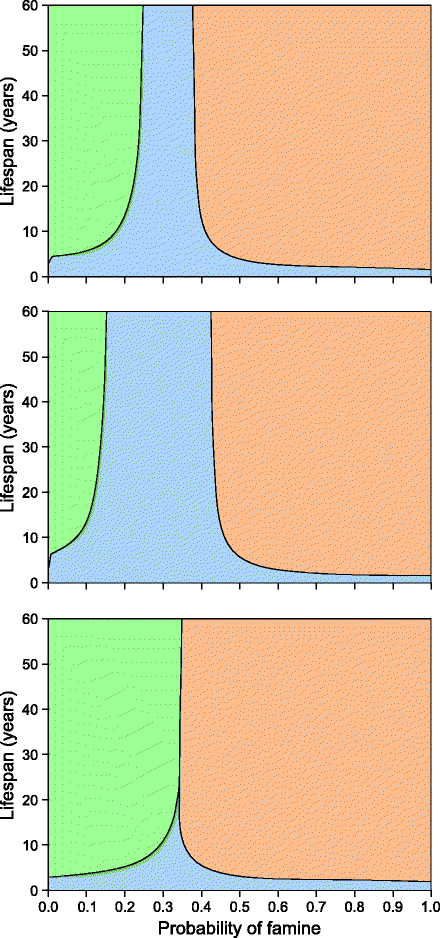
Fig. 4.3
Parameter areas of advantage of the three genotypes when there is a correlation between birth time and lifetime conditions (a) r = 0.1, (b) r = 0.2, and (c) r = −0.05. With a small positive correlation, the advantage of fetal programmer increases substantially. However, even very weak negative correlations can drive fetal programmer to extinction when life expectancy is high. Selection for fetal programming therefore must be driven by factors that produce significant positive birth time and lifetime correlations. Without such correlations predictive fetal programming is unlikely to evolve
Table 4.1
Correlations of annual rainfall to that of the subsequent year, short-term (10 years) and long-term (40 years) cumulative
S. no. | Subdivision | 1 year | 10 years cumulative | 40 years cumulative |
|---|---|---|---|---|
1 | Assam Meghalaya | 0.047 | 0.042 | −0.029 |
2 | Nagaland, Manipur, Mizoram, and Tripura | 0.006 | 0.010 | −0.078 |
3 | Sub-Himalayan West Bengal | −0.061 | −0.103 | −0.023 |
4 | Gangetic West Bengal | −0.009 | 0.096 | −0.265a |
5 | Orissa | −0.111 | 0.135 | −0.096 |
6 | Jharkhand | −0.054 | −0.042 | −0.012 |
7 | Bihar | 0.070 | −0.158 | −0.048 |
8 | East Uttar Pradesh | 0.096 | −0.106 | −0.091 |
9 | West Uttar Pradesh plains | −0.036 | 0.010 | −0.066 |
10 | Haryana | −0.010 | 0.050 | 0.041 |
11 | Punjab | −0.050 | 0.099 | 0.081 |
12 | Rajasthan | 0.048 | −0.200a | −0.094 |
13 | East Rajasthan | 0.047 | −0.034 | −0.017 |
14 | West Madhya Pradesh | 0.052 | 0.114
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|