(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
So far I have been arguing at the level of ultimate reasoning to address the question why physical activity, aggression, and other components of the soldier behavior syndrome should have a negative relationship with components of the insulin resistance syndrome. Now we need to look at the proximate mechanisms which execute this relationship. In order to understand the mechanisms and pathways involved in this connection, we need to know the physiology of aggression–dominance syndrome first.
So far I have been arguing at the level of ultimate reasoning to address the question why physical activity, aggression, and other components of the soldier behavior syndrome should have a negative relationship with components of the insulin resistance syndrome. Now we need to look at the proximate mechanisms which execute this relationship. In order to understand the mechanisms and pathways involved in this connection, we need to know the physiology of aggression–dominance syndrome first.
In traditional thinking testosterone has been associated with aggressive behavior. Testosterone is believed to facilitate aggression, and there is sufficient evidence to support this. It is predominantly a male aggression hormone. Since, generally, aggression research is male biased, the importance of testosterone appears to be overplayed. It also needs to be appreciated that the role of testosterone in aggression is contextual. A high-testosterone individual is not always aggressive but rather is more likely to be aggressive in certain contexts. It is much more important to realize that it is not testosterone alone that modulates aggression. Associating testosterone with aggression is a sign of a typical old-fashioned reductionism in biology. Today we have better tools with which we can understand the wide array of molecules involved in aggression. For example, Chiavegatto [1] lists knockout mouse models with altered aggression. This list has 36 genes and is by no means a complete list of genes involved in aggression. The list of knockouts with altered aggression illustrates a number of things that were not appreciated earlier. For example, not only androgen receptor mutants lose aggression, estrogen receptor α mutants and aromatase (the enzyme that converts testosterone to estradiol) mutants also lose aggression indicating an important role for estrogens in mediating aggression as well.
In addition to gene knockouts, another source of data is studies on gene expression in animals selected for aggression versus that in wild types. A pleasant surprise here is that there are some data on maternal aggression in females too [2]. These studies tell us that there are a large number of players with different roles in the physiology of aggression, and the testosterone-centered picture is too narrow and piecemeal. Maternal aggression, for example, is not chiefly testosterone driven but has been demonstrated to have the same beneficial effects against at least some of the components of metabolic syndrome such as hypertension [3]. Nevertheless, since there is a large volume of work on testosterone, we will have to continue referring to it every now and then, but we need to understand that it is not the only aggression hormone and also that aggression is not a male-specific behavior.
The pattern I discovered while scanning the literature on aggression was that majority of the aggression-associated molecules are also associated with metabolic syndrome in some way or the other. For example, out of the 36 genes listed by Chiavegatto [1], 28 are connected with metabolic syndrome in some way or the other. The total number of signal molecules that link aggression to metabolic syndrome is about 70, and the list is growing. Here I include hormones, neuropeptides, growth factors, and transcriptional regulators as well as dietary signals under the generic term “signal molecule.” For interested readers a complete list of these signal molecules with their nature of association with aggression and with metabolic syndrome with select references is compiled in Appendix I. The width of this association by itself is astonishing. It is difficult to believe that this is nothing more than a coincidence. It certainly points to a highly evolved link between the aggression pathways and the endobolic (i.e., endocrine and metabolic) pathways related to metabolic syndrome. Unless this association has evolved over the millennia, it is unlikely to involve such a wide array of links. Perhaps it is possible to find the evolutionary origins of this link. So far I have been unable to gather sufficient data to trace the origin, but there are indications that it is very old and conserved. We find this link in Drosophila, for example, which generally is a nonaggressive species. Fruit flies do not have any lethal weapons, and therefore, their fights are never fatal. Nevertheless, mild male–male aggression is observed during courtship over access to the females. A gene called sugarbabe which downregulates insulin production in the fly is overexpressed during courtship behavior in the fly [4]. Since in these flies there is little aggression apart from courtship, it is difficult to separate aggression from courtship. It is very likely therefore that sugarbabe overexpression is actually linked to courtship-related aggression. This raises a possibility that the connection between sex, aggression, and insulin may be very ancient and has been conserved during evolution. This needs to be thoroughly investigated.
Belsare et al. [5] showed that most molecules associated positively with aggression have a negative association with metabolic syndrome, and the ones negatively associated with aggression have a pro-obesity or pro-insulin-resistance action. This reverse association is highly significant. However, although I was a contributor to this paper, I now think that the categorization of positive and negative associations with aggression is too naïve. In complex control loops it is difficult to assign positive or negative signs to associations. For example, if A directly upregulates B and B through a negative feedback loop controls A, do we call the association of A and B as positive or negative? Endorphins or brain-derived neurotrophic factor (BDNF) are related to aggression in a negative feedback loop as we will see a little later. Aggressive encounter upregulates endorphin or BDNF, but both of them control aggression. This relationship is therefore positive in one direction and negative in the other. Until the exact role of a signal molecule is elucidated sufficiently well, it is difficult to infer anything from positive and negative associations. A number of molecules in the aggression regulation circuits have such relationships as we will see soon. Therefore rather than looking at positive and negative associations, it is better to look at the specific roles of each molecule and interpret them in the right context which I will attempt to do in this chapter. Although at this stage we do not know the specific role of every molecule, the fact remains that a large number of molecules appear to link aggression with components of the metabolic syndrome in some way or the other, and therefore, it is not possible to understand metabolic syndrome without understanding aggression.
The Bridge Between Aggression–Dominance and Metabolic Syndrome: Some Important Pillars
A. Serotonin: The endocrinology of territorial aggression and dominance in animals is well known. Perhaps the single most important molecule central to the suppression of aggression is serotonin in the brain as we have seen in the previous chapters [6–14]. Dominant individuals have low serotonin as compared to the subordinate ones among animal societies [15–19], and an intervention to raise brain serotonin activity in a dominant individual changes its behavior to that of a subordinate [15, 20, 21]. The arrow of causation appears to be dual, i.e., change in serotonin levels affects aggression and aggressive behavior modulates serotonin levels [6, 10]. In humans low serotonin activity is associated with impulsive aggression or violent suicide [8, 16, 22].
Serotonin is also central to metabolic syndrome in a number of ways. The relationship of serotonin with obesity is complex. Serotonin works as a satiety signal and thereby controls hunger. Therefore serotonin action should be antiobesity. Knocking out or blocking serotonin receptors leads to obesity as expected [23, 24]. Serotonin reuptake inhibitors have been proposed as potential antiobesity drugs, and some serotonin reuptake inhibitors have been shown to reduce body fat in short-term studies [25–27]. The effect, however, may be reversed in the long run. Many of the serotonin reuptake inhibitors that raise the standing serotonin levels lead to a paradoxical weight gain and insulin resistance [28–30]. It is possible that serotonin has an antiobesity action in the short run, but chronic elevation of serotonin gives rise to a state that can be called serotonin resistance. The concept of serotonin resistance is used in a slightly different context [31] but is similarly applicable here. Serotonin resistance may lead to obesity and insulin resistance. This is supported by data that demonstrate association of low serotonin responsiveness with insulin resistance [32, 33]. Chronically elevated levels of serotonin in the ventromedial hypothalamus have been shown to induce insulin resistance [34]. Serotonin reuptake inhibitors which increase basal serotonin levels are shown to induce insulin resistance in vitro [35]. Chronic signaling by serotonin along with norepinephrine in the ventromedial hypothalamus increases glucose sensitivity of the pancreatic β cells [36] and thereby increases the production of insulin. Reciprocally hyperinsulinemia and insulin resistance result in elevated serotonin levels [37]. Similar to insulin, leptin also increases serotonin turnover in the brain [38]. Thus there appears to be a mutually supportive mechanism. High levels of insulin and leptin which mark insulin resistance upregulate the serotonin levels, and chronically high serotonin induces insulin resistance. This is a typical positive feedback cycle that stabilizes a state. Serotonin reduces bone mass and strength on the one hand [39–42] and increases memory and cognitive functions on the other [43–48] with the exception of spatial working memory [47, 49, 50]. This is compatible with a warrior to diplomat transition. Serotonin also decreases sexual motivation and function [51, 52]. Thus serotonin seems to do almost everything that happens in the early insulin resistance syndrome and related conditions.
I must admit here that the picture painted above is rather oversimplified. Serotonin has a number of receptors having partly overlapping and partly differential functions. Science is just beginning to understand the roles of different serotonin receptors under different sets of conditions. Therefore a comprehensive picture of the network of effects of serotonin signaling is yet to emerge. Nevertheless, a robust message that everyone will agree with is that it is essential to better understand the aggression suppression hormone serotonin in order to gain sufficient insight into the insulin resistance syndrome.
B. Dopamine: The relation of dopamine with aggression appears to be diametrically opposite to that of serotonin. Dopamine activity is positively associated with aggression, and the arrow of causation also appears to be dual [6, 22, 53]. Dopamine in contrast to serotonin facilitates physical strength. The deletion of DAT gene (dopamine transporter) results in osteopenia and deficiencies in skeletal structure and integrity illustrating the role of dopamine in strengthening the bone [54]. This is compatible with dopamine’s proaggression role. It is no surprise then that dopamine has antiobesity and antidiabetic action. In mice, the use of dopamine receptor antagonist is associated with weight gain and insulin resistance, whereas agonists of the D1 and D2 dopamine receptor isoforms decrease food intake and improve insulin sensitivity [55, 56]. Dopamine may be extremely important in the chain of pathways leading to diabetes since diabetes is associated with decreased dopamine activity [57]. Dopamine, in contrast with serotonin, is crucial in spatial learning and memory [47, 58–60]. It is not difficult to visualize that spatial learning and memory are of critical importance to a hunter or warrior. Therefore a proaggression signal enhances spatial memory, whereas an aggression suppression signal enhances verbal memory, which is a diplomat requirement. Therefore serotonin which is a general memory enhancer does not boost spatial memory [47, 49, 50].
C. Cholesterol: Perhaps next to serotonin, cholesterol appears to have a major role in the suppression of aggression. The negative association between cholesterol and physical aggression is quite robust [61–64]. In contrast to physical aggression, nonphysical aggression has a positive association with cholesterol [65]. Cholesterol seems to have not only an association but a causal role in aggression control since cholesterol lowering appears to increase aggression [66–69]. But unlike serotonin, the mechanism by which cholesterol affects behavior is largely unknown. Cholesterol apparently does not cross the blood–brain barrier but still affects serotonin activity in the brain [70, 71]. This has been claimed as an adaptive response under famine conditions where a famine-induced lowering of cholesterol would lower serotonin activity allowing aggressive behavior that might be needed under intense competition for food [72]. But food and fat intake are not the only factor determining the blood cholesterol levels. There appears to be a CNS-mediated control on cholesterol too [73, 74]. It is likely therefore that there are behavioral inputs in the regulation of cholesterol, but not much research has gone into this aspect of cholesterol. There is no need to reemphasize the role of cholesterol in metabolic syndrome. Cholesterol strongly links aggression suppression to metabolic syndrome, but much research is needed to elucidate its precise role in both.
D. Corticosteroids: Similar to cholesterol, low cortisol levels are associated with aggression [75–78] raising the possibility that cortisol may serve as an aggression control signal. This interpretation is muddled by the concept of it being a “stress” hormone. We will devote a separate chapter to discuss the concept of stress, and therefore, I will not discuss it here except a passing remark that I think aggression suppression and risk avoidance are the major roles for which the corticosteroid response has evolved. I will discuss evidence for this statement in relation to stress more elaborately later since it deserves much more space. Cortisol is known to induce insulin resistance [79, 80] and therefore is another potential link between aggression suppression and insulin resistance.
E. Sex hormones: Sex and aggression are positively associated in a wide variety of species including vertebrates and invertebrates [81–83], many sex hormones serving as aggression hormones simultaneously. Dominant and physically aggressive individuals have high testosterone/estradiol levels [84–88]. Testosterone’s role in male aggression is well recognized. However, endocrinology of female aggression is not very clearly elucidated. Aggression is equally important in female life as well, although the context, nature, and intensity of aggression could be different from that of the males. There are certain female-specific forms of aggression such as maternal aggression. Unfortunately aggression research is largely centered on male aggression. I will have to repeatedly talk about testosterone in this as well as the following chapters, and contextually it would be more applicable to males. This is a bias that I do not like to have, but it is inevitable with the currently available data. I hope to see more research in the female-specific forms of aggression which would help us remove this bias eventually. Interestingly, in a species of mouse that shows biparental care, males show paternal aggression similar to the maternal aggression in females with much overlap in the mechanisms involved [89]. This could be particularly relevant to the human species which is characterized by biparental care. It appears that the importance of testosterone in aggression is overplayed at the cost of a number of other molecules involved in aggression in both genders. For example, similar to androgen receptors, estrogen receptor knockout mice also lose aggression [1]. But the role of estradiol in aggression is not as clearly recognized as that of testosterone. Nevertheless, there is evidence for an important but complex role of estradiol in aggression [90–95]. Apart from testosterone, dehydroepiandrosterone (DHEA) is an adrenal androgen precursor and is important for the expression of animal aggression in nonbreeding season when gonadal testosterone synthesis is low [96].
Testosterone facilitates glucose transport in muscles [97], reduces oxidative stress and accompanying cell damage [98, 99], and stimulates angiogenesis and vascular regrowth [100]. The multiple health beneficial effects of testosterone have given rise to suggestions that testosterone deficiency may be the prime factor behind metabolic syndrome, although the causal role of testosterone is debated [101]. Estradiol is also shown to promote growth and angiogenesis [102]. Estrogens, especially 17 β-estradiol and estriol, themselves are naturally occurring antioxidants [103]. Owing to their multiple actions, both male and female sex hormones have protective effects against metabolic syndrome in the respective sex [87, 104], although they may have different effects in the other sex [104]. DHEA is also protective against obesity, insulin resistance, and some of the mechanisms of diabetic complications [105–111].
F. Insulin: It is likely that hyperinsulinemia and insulin resistance itself are involved in aggression control. Parameters of insulin sensitivity were shown to be positively correlated to violent suicide [112], an association similar to low cholesterol. Since insulin enhances serotonin response, the aggression suppression action of insulin is likely to be mediated through serotonin. Insulin has a complex regulatory role in aggression, impulsivity, restlessness, and other behavioral characteristics [113], which appears to operate through the effects of insulin on the monoamine dynamics in the brain [114–118]. This role of insulin in the brain monoamine dynamics has been realized recently, and a clear picture is yet to emerge. But we know now that insulin substantially modulates monoamine actions which are important behavioral regulators. I suspect that insulin has a direct role in the suppression of anger, aggression, and impulsivity and hope that this is clearly demonstrated in the near future. On the other hand insulin has a direct positive role in cognitive functions of the brain [119–124].
G. IGF-1: A significant positive association has been shown between IGF-1 mRNA expression and the level of aggression in fish [125]. Dominant pudu males had higher IGF-1 levels than their subordinate pen mates [126]. IGF-1 expression is shown to be suppressed in subordinate baboons [127]. This dominance–aggression-associated molecule has a protective effect against obesity and insulin resistance. IGF-1 reduced hyperphagia, obesity, hyperleptinemia, and hyperinsulinemia in rats [128, 129]. Liver IGF-1-deficient mice showed insulin resistance [130]. IGF-1 also has vascular protective effects [131].
H. Cholecystokinin (CCK): Higher expressions of CCK and CCK2R were associated with aggression in transgenic mice overexpressing progastrin [132]. In normal mice brain, CCK levels elevated following aggressive encounters in adults or a rough and tumble play in adolescents [133]. The antiobesity and antidiabetic effects of CCK are well demonstrated [134–136]. CCK promotes growth of endocrine pancreas [137] and enhances wound healing [138].
I. Ghrelin: Ghrelin levels are negatively associated with cholesterol, and violent individuals have low cholesterol and leptin along with elevated ghrelin [139]. Although ghrelin is known to enhance appetite [140, 141] and thereby expected to increase obesity, low plasma ghrelin levels are associated with type 2 diabetes, insulin resistance, and hypertension [142, 143].
J. EGF, NGF and other growth factors: Aggression stimulates salivary epidermal growth factor (EGF) secretion [144–146]. Testosterone induces EGF synthesis but not secretion [147, 148]. EGF release is under autonomic nervous control [148–150]. Therefore a combination of testosterone and sympathetic stimulation, a characteristic of aggressive behavior, is necessary for EGF release. The adaptive logic behind why salivary glands are the major sources of EGF and why aggression stimulates their secretion is that aggression anticipates injuries and EGF is needed in wound healing. Since animals lick wounds, salivary glands are the best places for the secretion of these growth factors. In T2D, there is a deficiency of EGF [151–153] which is likely to be due to deficiency of aggressive behavior. This is one of the reasons for delayed wound healing in diabetes. Apart from wound healing, EGF has an important role in β cell regeneration too [154], and patents have been filed for therapeutic use of EGF in stimulating β cell growth [155]. EGF also plays a role in retinal and neuronal integrity [156, 157]. The aggression-stimulated EGF thus has multiple roles in protecting against diabetes.
Similar to EGF, the secretion of nerve growth factor (NGF) is also stimulated by aggression [158–163] or adventure [164]. Apart from a general role in wound healing and tissue regeneration, NGF has specific roles in maintaining nerve integrity as well as retinal integrity, and deficiency of NGF is one of the suspected causes of diabetic neuropathies [165, 166] and retinopathy [167].
Apart from EGF and NGF, a number of other growth factors play a role in aggression, injuries, and β cell regeneration that have not attracted sufficient research as yet. They include platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), growth hormone (GH), prolactin, placental lactogen, parathyroid hormone-related peptide (PTHrP), gastrin, gastrin-releasing peptide (GRP), glucagon-like peptide, and betacellulin. As we will see more elaborately in a later chapter, many of them are involved in wound healing and are synthesized locally or systemically after even minor injuries. Interestingly most of the growth factors needed for wound healing are also involved in β cell regeneration. This is a factor linking aggression–dominance syndrome to diabetes in a different way. Hawk or warrior individuals are more prone to injuries, and owing to the fitness importance of injuries, we can expect evolution to have built-in upregulation of wound healing mechanism on adopting aggressive behavior. Since a number of growth factors are common, this upregulation could also strengthen endocrine pancreas. A deficiency of aggressive behaviors is likely to lead to a cumulative deficiency of growth factors and thereby affect β cell function. In addition to helping β cell regeneration, some of the growth factors have been shown to exert systemic anti-inflammatory action [168].
K. BDNF: The BDNF bridge between aggression and diabetes appears to be built in a different way. Frequent aggression [169] and particularly repeated defeat in fight appear to upregulate BDNF [170], and BDNF controls aggression. BDNF deficiency leads to aggressive phenotype [171, 172]. There exists a link between EGF, NGF, and BDNF such that EGF and NGF together enhance BDNF activity [173]. The role of BDNF in diabetes is that it is insulin sensitizing [174] and has a protective role against neuropathies [165, 175]. BDNF heterozygous knockout mice are obese and insulin resistant [176]. BDNF levels are reduced in diabetes [177] and Alzheimer’s disease [177]. Unlike all the bridges listed above, BDNF is an anti-aggression molecule and still has a protective role against diabetic complications. Does this go against our hypothesis? We will soon see that it does not. But to understand that argument, we need to understand first the socioecophysiology of aggression in a holistic way.
L. Endorphins: The brain opioid peptides including endorphin are aggression control signals [178, 179]. It has two apparently contradicting links with obesity and insulin resistance. On the one hand it facilitates overeating and is positively associated with obesity and insulin resistance [180–183]. On the other hand it is involved in exercise-induced insulin sensitization [184, 185]. This apparent contradiction will also get resolved when we will see the socioecophysiology of aggression.
M. Gamma-aminobutyric acid (GABA): The role of brain GABAergic activity in aggression and impulse control is similar and perhaps synergistic to that of serotonin [186–189]. Although GABA is involved in aggression control, the knockout of GABA-synthesizing enzyme GAD65−/− is paradoxically nonaggressive. This paradox will also get resolved when we understand aggression holistically. GABA is also intricately involved in food intake regulation and thereby obesity [190–195].
N. Growth hormone (GH): GH administration increases isolation-induced aggression [196–198]. GH is essential in building up muscle mass, although its contribution to muscle strength is debated [199–203]. In obese individuals the GH response is blunted [204], and GH deficiency is characterized by central obesity. The growth-promoting action of GH is in synergy with the action of insulin, and therefore, it is expected that GH would upregulate insulin secretion, which may be accompanied by increased resistance. The action of GH does increase insulin resistance [205, 206]. However, the GH signaling pathway involves IGF-1 which is insulin sensitizing [129, 130, 207], and therefore, the net effect of GH would be enhancing insulin action.
O. Progastrin–gastrin: Transgenic mice overexpressing progastrin showed increased aggression [132]. On the other hand central obesity and insulin resistance were increased in mice lacking gastrin expression [208]. Gastrin in combination with EGF stimulates β cell regeneration and therefore has antidiabetic action [154, 209].
P. Myostatin: Myostatin limits muscle growth, and increasing levels of myostatin are thought to be responsible for age-induced sarcopenia. The aggression hormone testosterone suppresses myostatin [210] and thereby promotes muscle growth. Myostatin sets the limit to muscle growth [211] presumably by downregulating mTOR pathway [212]. Along with downregulation of mTOR, myostatin increases insulin resistance [213], and myostatin inhibitors, known to facilitate muscle growth, prevent the development of obesity and type 2 diabetes [214]. Myostatin thus mediates a negative association between muscle strength and insulin resistance.
Q. Nitric oxides (NO): Nitric oxides and the NO synthases have complex involvement in both aggression and metabolic syndrome. NO signaling is increasingly known to be involved in diverse functions of the body. Reduced bioavailability of NO characterizes obesity and related disorders [193]. Interestingly endothelial NO synthase (eNOS) and neuronal NO synthase (nNOS) have different effects on aggression and accordingly have differing effects on metabolic states too [10, 215–224]. The eNOS is proaggression and insulin sensitizing, whereas nNOS has mixed effects on aggression and also mixed effects on insulin resistance.
R. Phosphoenolpyruvate carboxykinase (PEPCK): PEPCK is a key enzyme in glucose metabolism and has an interesting relationship with insulin resistance and with aggression. Overexpression of this enzyme in liver leads to liver insulin resistance and upregulation of gluconeogenesis [225, 226]. Its overexpression in adipose tissue leads to increased accumulation of fat. This kind of obesity is not accompanied by higher levels of FFAs in blood. Also in spite of obesity, there is no change in insulin levels or insulin sensitivity [227]. This demonstrates that if only fat deposition is specifically stimulated without other accompanying changes, insulin resistance does not follow, implying that some other process accompanying development of obesity could be more important for insulin resistance than obesity itself. More interesting is the case with muscle. If this enzyme is engineered to overexpress in muscle, it increases endurance making the muscle almost immune to fatigue. Interestingly animals with such increased muscle strength increase their aggression levels substantially. These aggressive animals have high levels of intramuscular triglycerides, but they are highly insulin sensitive [228]. PEPCK overexpression in muscle is not a naturally occurring condition, and therefore, it may not be of any clinical relevance. But it gives us an important insight into the physiology of aggression as well as the origin of metabolic syndrome, which we will see shortly.
S. Mammalian target of rapamycin (mTOR): The mTOR is a protein in the downstream pathway of insulin or IGF-1 and IGF-2 and regulates cell growth. Increased mTOR activity reduces insulin action which works like a feedback regulator of insulin action [229–231]. The mTOR signaling pathway is activated by excess nutrients such as FFAs or amino acids and insulin itself. This is likely to be the mechanism of diet-induced insulin resistance. Therefore mTOR is thought to be a crucial link in insulin resistance. Interestingly mTOR is activated by exercise [232–236] and testosterone [237, 238], but both exercise and testosterone are insulin sensitizing. This means that physical activity and testosterone, both important components of physical aggression, increase the mTOR threshold for insulin resistance. There is evidence for interaction between testosterone, insulin, and mTOR pathway [239]. mTOR is involved in protein synthesis and cell growth, which is particularly important in strengthening muscle. The action of myostatin on mTOR appears to be opposite that of testosterone and exercise. Myostatin suppresses mTOR but induces insulin resistance. So fine-tuning of mTOR pathway could be the key link between aggression, muscle strength, and insulin action.
T. Sympathetic activation components: What is perhaps the most important mechanism linking aggression to obesity is the interplay between sympathetic nervous system, adipose tissue, and testosterone. Visceral adipose tissue is innervated by autonomic nerves. Sympathetic activation of the adipose tissue is long known to have a lipolytic action. This action is through β adrenergic receptors, and testosterone enhances the expression of β adrenergic receptors on adipocytes [240, 241]. Therefore when testosterone levels are adequate, with every environmental and social stimulus that leads to sympathetic activation, there is lipolysis. However in testosterone deficiency, typical of nonaggressive personality, lipolysis is arrested. A combination of testosterone and sympathetic actions, a characteristic of aggressive encounter, prevents accumulation of visceral fat [242–246]. The other effects of sympathetic activation including liver glucose production are not affected by low testosterone. In effect, sympathetic activation may release more glucose for lipogenesis. Thus, in the presence of testosterone, sympathetic activation has antiobesity effect, but in its absence, the same sympathetic stimulation may contribute to fat accumulation.
U. Osteocalcin: The sex and aggression signals is an adjective to testosterone and estradiol to facilitate osteocalcin production [247–251]. A marker of bone formation and strength, osteocalcin induces β cells to release insulin and also induces the release of adiponectin [248] which has a strong antiobesity, anti-inflammatory, and insulin-sensitizing action. Osteocalcin also has insulin-sensitizing and antiobesity actions [248–252].
V. Oxytocin: Oxytocin has a somewhat mystic role in the network of links between aggression and metabolic syndrome. The behavioral effects of oxytocin are increasing trust and affection on the one hand [253–261] and increasing aggression on the other [255, 262–266]. As an expected corollary, the effects of oxytocin on obesity, hypertension, and insulin resistance are also mixed: It facilitates lipogenesis but has antiobesity effects, and it facilitates liver gluconeogenesis yet reduces insulin resistance [257, 258, 267–277]. This needs careful interpretation, but as you will see below once we understand the ecophysiology of aggression in a broader perspective and place oxytocin in its natural role, the apparent contradiction gives way to logical coherence.
W. Glucose: Low levels of plasma glucose are long known to stimulate aggression [278–280]. Anthropologists have discussed and debated over the hypoglycemia–aggression hypothesis explaining the differences in aggressive behaviors within and between ethnic groups [281–283]. Close to the physiological range, plasma glucose is negatively related to aggression. Low glucose levels promote aggression, but unusually low glucose levels arrest aggression.
X. Tryptophan and arginine: Tryptophan is one of the fewer clearly known links between diet and aggression. Tryptophan supplementation reduces and depletion increases aggression [13, 20, 284–288]. This is because tryptophan is a precursor of serotonin in the brain and can pass the blood–brain barrier. Arginine is another known amino acid bridge between insulin and aggression. Although a number of amino acids stimulate insulin release, individual amino acids differ in the elicited response. Arginine has a specific insulin secretion stimulatory effect [289–291]. Arginine reduces aggressive response [292] as expected for an insulin-secreting agent.
Y. Vitamin D: Vitamin D is an important essential vitamin that is known to be important for bone strength and integrity. Disruption of the vitamin D receptor gene results in loss of aggression in mice [293] accompanied by an increase in grooming behavior [293, 294]. Compatible to our hypothesis, vitamin D deficiency is identified as a significant contributor to insulin resistance [295], and vitamin D3 supplementation is shown to be an effective insulin sensitizer [296].
Z. Fat and triglycerides: Plasma triglyceride levels negatively correlated with aggression [297] consistent with the hypothesis. Leptin levels which are positively correlated with fat are negatively associated with aggression [69, 298]. Monkeys fed with a high-fat high-cholesterol diet were observed to be less aggressive than those on low-fat low-cholesterol diet [71, 298, 299]. This is perplexing at a glance since both fatty acids and cholesterol do not cross the blood–brain barrier to reach the brain and exert any behavioral effects. An interesting mechanism is suggested as to how fat diet may reduce aggression [69, 112, 300]. Fatty acids compete with tryptophan for albumin binding. As a result when FFAs are high, more tryptophan remains in the free state. Since only free tryptophan can pass the blood–brain barrier, more of it is available in the brain when plasma FFAs are high. Tryptophan is a precursor of serotonin, and its concentration can be a limiting factor in serotonin synthesis. Therefore high levels of FFAs in plasma can increase serotonin synthesis in the brain without crossing the blood–brain barrier. Although speculative at this stage, the hypothesis is promising and may be a strong link between diet and behavior. FFAs therefore are important molecules bridging behavior and biochemistry. It is likely that many other dietary factors influence behavior, but research in this direction is scanty.
This list, although A–Z coincidentally, is not really the A–Z of the links between aggression and metabolic syndrome. This is only an incomplete list of molecules associated with aggression and injuries that are also associated with metabolic syndrome but includes the ones about which there are sufficient data so that we can appreciate their relevance to metabolic syndrome. For example, the peptide proopiomelanocortin (POMC) and its cleavage products are intimately involved in aggression control as well as in obesity and insulin resistance, but the POMC system by itself is so complex that we may have to wait for a few more years’ research to make logical sense out of all the components of POMC and their interactions. Nevertheless, there are clear indications that POMC and component peptides are very important links between aggression and obesity. The total number of links between aggression and metabolic syndrome is much greater (Appendix I) and will probably need another few decades of research to understand them in a coherent and comprehensible way. Here I need to restrict myself to less than half of them. Having looked at the role of some of the individual molecules, we need to join the pieces together to make a coherent picture, although the picture will be far from complete at this stage.
Often when pieces of a complex system are joined together, some emergent properties are seen which are not displayed by any of the components themselves. In fact, such emergent properties are at the core of biological systems. Therefore we need to look at the aggression–dominance syndrome in the behavioral, ecological, and neurophysiological context at the same time. This is particularly important. Aggression has been addressed by behavioral ecologists, psychologists, and neurophysiologists for a long time. But each group has created its own niche, and they rarely come out of their expertise castles to exchange ideas. Therefore a holistic picture of aggression has failed to emerge. Since I am not an expert in any of these fields, I can comfortably try to combine the piecemeal research on aggression and try to construct a coherent picture.
The Complex Physiology of Aggression
Physical aggression is an event that has important lifelong consequences. The reward as well as the risk levels of aggression are very high, and therefore, natural selection must have fine-tuned all the mechanisms involved. The decision to initiate aggression or retaliation is an extremely important decision, and a wrong decision can cost life. Therefore mechanisms must have evolved to check and countercheck the decision before implementing it. Before initiating an aggressive act, it is necessary to check that all mechanisms needed for effective implementation of aggression as well as for coping with possible consequences are functioning well. If one or more of them are likely to fail, it is better to avoid aggression. This I called as the “aggression checkpoint” in the last chapter. Let us see now what the essential requirements for execution of aggression are and accordingly what the essential checkpoints are.
It is obvious that bone and muscle strength is needed for effective physical aggression. This is evident by the loss of aggression and increased submissive behavior associated with vitamin D deficiency which weakens the bone architecture. It is crucial that some mechanism for assessing muscle strength and muscle energy supply should exist, and they must feed this information into the decision-making system for initiating aggression. Not much research has addressed this question directly, but there are indications that this could be happening. Everyone is familiar with the feeling of fatigue or weakness when muscle energy sources are depleted. This feeling makes aggression less likely. It is possible that the sensory nerve endings in muscle that are known to be stimulated by every contraction continually keep on collecting this information. Myokines that are released with every muscle contraction are also likely signal molecules. This is currently speculative. But a recent piece of experiment demonstrates how muscle energetics influences aggression. When experimenters engineered overexpression of PEPCK in muscle, the muscle energy metabolism received an extra boost and became almost immune to exhaustion. This was demonstrated by the sustained hyperactivity of these animals. The most interesting point to be noted is that these animals became highly aggressive. The experimenters had manipulated only the energy supply to muscle and nothing else. There was no primary neuronal or hormonal change that could have resulted in aggression. Since only increasing muscle energy increased aggression, it suggests that there must be some mechanism that constrains aggression based upon muscle capacity. Muscle capacity must have been an important limiting factor in aggression, and when this limit was lifted, aggression control got lifted too. I feel confident in concluding that the checkpoint of muscle and bone strength/capacity must be an important controller of aggression. If this is true, it implies that any energetic disinvestment from muscle will control aggression. Insulin resistance or insulin deficiency results into energetic disinvestment from muscle, and therefore, it is no surprise that diabetes is accompanied by loss of aggression [301].
Strength is not the only requirement for successful aggression. Aggression has conflicting hemodynamic requirements. On the one hand there is explosive muscle action that needs increased blood supply. Vasodilatation is one of the mechanisms to increase the blood flow. On the other there is increased risk of injury, and in order to minimize bleeding, vasoconstriction is also adaptive. Therefore both vasodilatation and vasoconstriction mechanisms must be boosted for successful aggression. Nitric oxides are crucial in vasodilatation and endothelins in vasoconstriction. The fact that eNOS knockouts [302] as well as ET-1 knockouts [303] lose aggression illustrates that there exists a hemodynamic checkpoint too. The involvement of both NO and ET-1 in an aggressive act is downstream to decision making in the process of aggression. Still impairment in NO or ET-1 dynamics inhibits aggression. Perhaps similar is the case with adrenalectomy which results in loss of aggression [304]. Aggression induces salivary EGF production in anticipation of injuries. However, removal of salivary gland in mice results into a delayed suppression of aggression and enhancement of defensive behavior [305]. Extracts of salivary glands induce aggression [306] demonstrating that signals from salivary glands are involved in initiating aggression. Both salivary and adrenal hormones are downstream in the aggression pathway, but they are essential in starting aggression. Barring checkpoint it is difficult to think of any other explanation for a downstream factor arresting an upstream process.
For the success of an aggressive encounter and from the point of view of lifetime fitness of an individual, a decision to stop an aggressive act at the right time is as important as the decision of initiating it. Therefore before initiating aggression, there must be a checkpoint for mechanisms of stopping it. The decision to stop is mainly driven by impulse control mechanisms involving serotonin and GABA. It is interesting to note that some of the GABA-related knockouts GAD65−/− and serotonin-related knockouts 5-HT1A−/− and 5-HTT−/− lose aggression. This is apparently paradoxical. GABA and serotonin are involved in aggression control, so their elimination should increase aggression, but in reality, it is shown to decrease. This paradox is resolved by the concept of aggression checkpoint. If the mechanism to stop aggression is impaired, starting aggression can be counterproductive, and evolution is likely to have hardwired mechanisms of countercheck. Thus there are multiple evidences for aggression checkpoints, but we do not know as yet how exactly they work. This is mainly because the concept is new, and no research inputs have gone into this question as yet.
The situation at the beginning of an aggressive encounter is likely to change rapidly during the encounter. Accordingly the decision may have to be revised as well as reversed as per the need. These mechanisms also must have evolved since these are life and death decisions. If the fight is prolonged, at some stage muscles will get exhausted and one has to stop. However it is unwise to wait for complete exhaustion of energy. In most species intraspecific aggression is not fatal, it is meant to test the relative strength and endurance of the fighters. It would be adaptive then to fight till the last second possible. However, fight with a given opponent is not the only concern for survival. There are other elements including predators. If a predator is sensed immediately after a fight, there needs to be sufficient energy reserve to run for life. Therefore it is not good to fight till exhaustion of available energy. A stop signal must come much before actual energetic exhaustion. This function is served by the central fatigue signals which get activated and stop the aggressive act much before peripheral energy supplies are burnt out. Interestingly the central fatigue signals are also driven by serotonin and GABA. Since serotonin and GABA are negatively correlated with aggression, in a test of endurance, a less aggressive individual is likely to get exhausted first and withdraw from fight. A more aggressive individual is more likely to win. But here lies a trade-off. Aggressive individuals are more likely to win a given combat but would be left with lower energy reserves. As opposed to that in nonaggressive individuals, the basal serotonin and GABA signaling would be high, and therefore, the central fatigue mechanisms would stop physical activity at an earlier stage. This would burn less energy but increases the chances of a defeat. There is an interesting modern consequence of this mechanism. When nonaggressive individuals, i.e., ones with high levels of the central fatigue signals, try to exercise, they get premature fatigue, and the energy reserves are not burnt by exercise as much as expected. This could be one of the mechanisms by which loss of aggression can contribute to obesity.
Other signals of exhaustion include endorphins and cortisol. Exercise research has shown that physical activity increases endorphin and cortisol, but this increase is not linear. Both endorphin and cortisol are not primarily needed for physical activity or aggression. The mechanisms primarily needed such as catecholamines that ensure energy release increase linearly with exercise [307, 308]. Endorphin and cortisol, on the other hand, start increasing only after prolonged and exhaustive activity [307, 308], and they are actually meant for signaling exhaustion and controlling aggression. Therefore the anti-aggression activities of endorphin and cortisol are adaptive.
The conditions for initiating and arresting aggression need to be weighed against levels of desperation in the context of life history trade-offs. An individual with little energy reserve is more desperate for food and therefore should be more aggressive in competition. It is not surprising therefore that hunger signals such as ghrelin have proaggression effects. An individual with larger energy reserves can afford to wait for better and less competitive opportunities in the future and therefore retract from aggressive encounter earlier. Here again serotonin and endorphin appear to play a role. Leptin upregulates serotonin, and therefore, obese individuals would activate central fatigue signals and withdraw from aggressive encounter faster. FFAs also play a role in upregulating serotonin as seen earlier. Energy-depleted individuals have a reduced chance of long-term survival, and therefore, they should utilize the present more desperately rather than relying on the future. Therefore they are expected to take more risks, fight longer, and exhaust their energy reserves more. Although sounding somewhat paradoxical, it is adaptive for individuals with higher energy reserves to spend less energy and those with lower reserves to spend more in highly competitive situations.
Similar to exhaustion, aggression needs to be controlled after perceived defeat. Defeat increases the central opioid peptides [178, 309, 310]. Brain opioid activity decreases aggression [178]. This negative feedback loop appears to have evolved as an adaptive response. Proactive aggression will be counterproductive after exhaustion or in front of a stronger opponent, and therefore, aggression suppression mechanisms are needed along with suppression of pain. The defeat response suppresses pain as well as aggression. This appears to be mediated by brain opioids. BDNF works in a similar negative feedback loop but on a more chronic scale. Repeated defeats are needed for raising BDNF [170]. Cortisol after defeat will have a similar function of suppressing aggression and risk-taking behavior. Losing is not only about controlling aggression. There is a need to cope with other consequences of losing. There could be injuries, and both immediate and delayed strategies are needed to take care of the wounds. The acute requirement is to prevent bleeding, and vasoconstriction is important for this purpose. After withdrawing from aggression, vasodilatation is no more needed, but vasoconstriction is since bleeding would continue even after the end of the fight. Endothelin is an important player here. So the balance between NO and ET-1 would be tilted towards endothelin on perceiving defeat and shunning aggression. The HIIR state appears to be involved in shifting this balance [311]. Endothelin and insulin are in a positive feedback loop. Insulin signaling upregulates endothelin production [121, 312, 313], and endothelin induces insulin resistance [314–317] as well as insulin secretion [318]. This may be adaptive on the face of a possible defeat where it is necessary to suppress aggression and reduce peripheral blood flow in anticipation of injuries. It is not surprising then that endothelin levels are generally increased in obesity and diabetes [319–322]. It is no surprise therefore that aggression suppression is associated with hypertension [323, 324].
As opposed to the loser the winner has a different metabolic state. In a winner, testosterone and dopamine activities go up rapidly, and cortisol is lowered. Both testosterone and dopamine signal the capability and readiness for aggression in a future encounter. This again is a positive feedback loop in which a winner attains a hormonal and metabolic state that increases aggressiveness, mobilizes mechanisms of strengthening muscle and bone, and thereby increases the probability of winning in future. Loser, on repeated defeats, attains a hormonal and metabolic state that prompts submissive behavior and risk avoidance. This positive feedback has long-term consequences and is likely to give rise to sustained behavioral syndromes that we are referring to as hawk–dove or warrior–diplomat (Fig. 7.1).
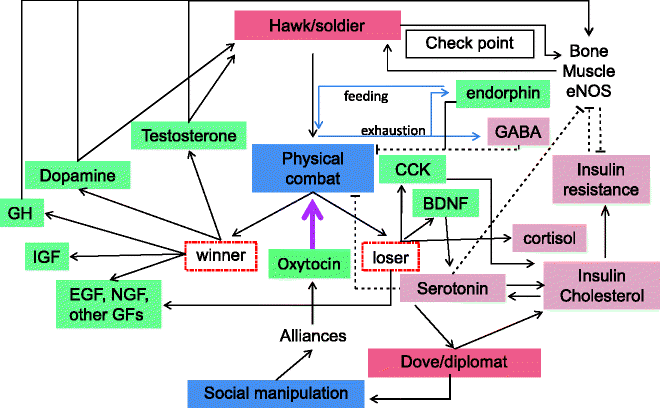
Fig 7.1
The physiology of aggression and its relevance to the insulin resistance syndrome. Pointed arrows on solid lines denote upregulation, and blunt arrows on dotted lines denote downregulation. There are two positive feedback loops that give rise to two different basins of attraction which are stable by themselves. Factors in green are known to have insulin-sensitizing, antiobesity, or antidiabetic effects. Factors in purple are known to be involved in the insulin resistance syndrome
The long-term sustenance of hawk or dove strategies is very different from a win–lose outcome in a short-term encounter. Acute and chronic suppression of aggression have different consequences, and it is a crucial point to realize. Aggression suppression due to exhaustion is temporary and can reverse quickly with replenishment of energy. In this context, it is appropriate that the exhaustion signal endorphin mediates hyperphagia in an attempt to restore energy [325, 326] but has insulin-sensitizing action on the background of recent physical activity. Aggression suppression necessitated by being substantially weaker than the opponent cannot and should not be reversed unless there is dramatic change in situation. The anti-aggression activity of exhaustion signals such as endorphin has evolved to stop aggression at the right time, whereas chronic aggression suppressors such as serotonin and cortisol are evolved to arrest initiation of aggression itself. The acute and chronic aggression suppression mechanisms are not two different mutually exclusive compartments but are two ends of a continuum. Some signals are involved in both, and some like BDNF appear to take an intermediate position. BDNF activity increases after repeated defeat [170]; BDNF is an aggression suppressor and has synergy with serotonin activity [327, 328]. However BDNF levels are not sustained on chronic aggression suppression. This is because BDNF is not a part of the positive feedback cycle of insulin resistance syndrome described below. As a result the initial phases of commitment to a dove or diplomat strategy might witness a high BDNF level which will be depleted eventually. Similar is likely to be the case with NGF, which is also not a part of the diplomat positive feedback circuit. Therefore a prolonged period of diplomat behavior and lack of soldier behavior would eventually lead to deficiencies of BDNF and NGF, although that may not be the case initially. In contrast with BDNF, the raised levels of serotonin and cortisol are sustained in chronic aggression suppression as they are a part of the positive feedback cycle, and they are the true stable makers of a diplomat mode of life. Similarly testosterone and dopamine may be sustained by a winner, and they mark a hawk or warrior strategy. Many other molecules upregulated in a defeat response do not sustain their raised levels chronically. These include EGF, NGF, and other growth factors. The autonomic nervous system also undergoes shift in activity. A sympathetic response is typically needed for aggression, whereas loss of aggression would lead to an inclination towards parasympathetic activity. Parasympathetic activity enhances pancreatic insulin response [329–331], and this may have a crucial role in maintaining the hyperinsulinemic state of a dove or diplomat. A hyperinsulinemic state needs to be necessarily accompanied by two compensatory changes, without which the organism will not survive. One is insulin resistance. Hyperinsulinemia without insulin resistance will lead to fatal hypoglycemia, and insulin resistance must accompany hyperinsulinemia to avoid this. A number of short- and long-term mechanisms exist that induce insulin resistance in response to hyperinsulinemia. The second response is that of disruption of food intake regulation mechanisms. Insulin is a potent regulator of food intake. If substantially raised insulin levels are sustained with normally functioning anorectic reflexes, the individual will almost never eat and die of starvation. In order to avoid this the appetite regulation mechanisms need to be disrupted, and evolution appears to have built-in mechanisms to do this as well. This can lead to type 1 thrift which is adaptive for a subordinate individual for reasons as under.
The hyperinsulinemic diplomat state has adequate provision of positive feedbacks to sustain itself. Hyperinsulinemia induces insulin resistance which results into disinvestment from muscle. The partially energetically deprived muscles give aggression suppression signals and prevent a transition back to aggression. Here lies the difference in the fine-tuning of strategies. If temporary exhaustion is the cause of aggression control which can be reversed after energy replenishment, disinvestment from muscle should not happen. This is ensured by the insulin-sensitizing action of the combination of exercise and endorphin. Thus short-term aggression suppression is prevented from getting into the vicious cycle. But if aggression suppression is chronic, it gets into the positive feedback cycle to stabilize the state. There is a positive feedback loop between insulin and serotonin as seen earlier which is stabilizing. The combined effect of serotonin and parasympathetic stimulation would raise insulin production. High insulin levels are adaptive for diplomat life since they enhance cognitive functions. Similarly leptin [332] and cholesterol are also needed for a similar purpose. But both these molecules have an anti-aggression effect further strengthening and stabilizing the state. High levels of insulin mediate insulin resistance and the chain of metabolic alterations downstream to it.
“Thrift” is particularly adaptive for the dove or subordinate life for multiple reasons. One is that subordinate individuals have less access to food and lower food security. Since dominant individuals can snatch food, the subordinates need to gulp as much as they can whenever it is available. Another reason is that since the chances of gaining immediate reproductive success are lower, they have to wait for a future opportunity. This necessitates investing in the future in the form of energy reserves. For this purpose hyperactivity of central fatigue signals is highly adaptive. Risk avoidance behavior in the present must accompany any strategy of investing in future. This role is mainly played by cortisol. Although energy intake may be increased, insulin resistance and cortisol arrest energy supply to muscle, and energy flow is diverted to lipid synthesis instead. Here lies a potential conflict of mechanisms, and evolution appears to have resolved that as well. Since leptin has important role in the cognitive brain functions, higher levels of leptin are needed. However, high leptin levels will prevent energy intake. This conflicts with the simultaneous requirement of thrift. The possible conflict appears to be resolved by evolution through mechanisms of leptin resistance. Leptin resistance allows hyperphagia and fat accumulation in spite of high leptin levels. There is some evidence that hyperinsulinemia and insulin resistance are sufficient to induce leptin resistance [333, 334]. Once leptin resistance develops, there is a clear path towards accumulating fat. Fat further reduces aggression by making more tryptophan available for serotonin synthesis completing one more turn of the cycle. These multiple positive feedback loops make the insulin-resistant state highly stable.
If both the aggressive hawk and nonaggressive dove/diplomat states are stabilized by positive feedbacks, is there any way to transit from one state to the other? There surely is. The transition from hawk to dove can be induced by repeated defeats, weakness due to any disease or major debilitating injury, or any other condition that makes aggression impossible for sufficiently long duration. Under such conditions it is adaptive to shun aggression for some time and focus on eating and resting to regain strength as much as possible. This marks a transition from hawk to dove state. In social species it is good for a weaker individual to be subordinate, groom other individuals, be more social, and make alliances. This may prepare one for getting back into the aggressive social rank struggle in the future. In social primates it is common for weaker males to make alliances and challenge the dominant alpha male. This strategy often succeeds. If the social strategies are successful, there can be a chance of reversal of the dove state. Mechanisms for this reversal also exist. For example, successful social relationships increase oxytocin which counteracts the effects of corticosteroids. After lowering corticosteroid activity, there can be greater risk tolerance, and slowly the behavior pattern may shift away from dove. Somewhat less known is the effect of oxytocin on protein synthesis [335]. By stimulating protein synthesis oxytocin could shift the metabolic bias from being lipid anabolic to protein anabolic, and this could be important in shifting behavioral strategies too. Getting back into the aggression cycle after making alliances is a tricky process. Stable alliances need trust and affection and utilizing the alliance to challenge a rival group needs aggression. Aggression and affection need to coexist during this transition, and this is appropriately achieved by oxytocin. Oxytocin has complex context-dependent behavioral effects, and it facilitates both affection and aggression [253]. Further oxytocin has complex effects on obesity and insulin resistance as well which are also likely to be context dependent. Our understanding of oxytocin so far is contradictory and confusing since evidence is contradictory. I think this apparent contradiction is an effect of trying to label oxytocin actions out of context. Oxytocin is a player on tricky grounds, so it needs to have evolved complex actions in combination with other hormonal and metabolic signals and under varied social and ecological conditions. Research now needs to focus on studying oxytocin action as a potential pathway for the transition from dove to hawk behavior using social alliances. In short, although both the aggressive and nonaggressive states are robust and self-supporting owing to stabilizing positive feedback loops, transition from one stable state to the other is possible under appropriate conditions, and pathways and mechanisms for both the transitions have evolved.

Here lies the difference between animal and modern human societies. In animals dove is frequently a reversible state, and for gaining higher ranking, one has to get back into physical dominance, although social alliances help in doing so. Since the dove state is reversible eventually, the metabolic state would also reverse substantially. In the modern human society, gaining higher status is possible, in fact easier, by being and remaining a diplomat. Therefore the need to reverse a diplomat state is hardly ever felt. It is interesting to note that the pathological effects of a diplomat metabolic state, i.e., an insulin-resistant state, are not observed for a long time. This is perhaps because we have evolved mechanisms to cope with this state for quite some time, until getting the right circumstances to reverse it. Basically the dove response has evolved as a transient adaptive state to reverse ultimately to the higher fitness-giving hawk state. This transition does not happen now with the modern human social structure, but we are still evolved for reversibility of the state after some interval. It is only after surpassing the time for which a diplomat state would be naturally sustained that we start observing the pathological effects. It is possible therefore that our diplomat state is not only quantitatively supernormal but is also temporally supernormal, and this supernormal temporal response leads to pathological consequences after several years.
The picture constructed above by joining several pieces of a jigsaw puzzle is a generalized common minimum principle. Although I expect it to be qualitatively similar and conserved, there would be subtle quantitative refinements in different species and different ecological setups as well as difference between individuals shaped by genetic, epigenetic, developmental, social, and other stochastic factors. These differences can be appreciated only when a common principle platform exists. Much more research is needed to understand how fine-tuning of these mechanisms takes place under varying conditions. Here I have only attempted to construct such a common platform in a brief comprehensive way. This should not be taken as an invariable necessity.
After having made a comprehensive sketch of the physiology of aggression in a behavioral ecology setting (Fig. 7.1), let us look at how it reflects on the clinical aspects of T2D and related disorders. It can be seen that almost all molecules that are acutely activated during aggressive encounter, irrespective of triumph or defeat, have antiobesity and antidiabetic action. These include sex hormones, dopamine, endorphins, BDNF, CCK, EGF, NGF, and other growth factors. Also markers of sustained hawk strategy including testosterone and dopamine are insulin sensitizing. Only the chronic dove or diplomat state markers such as serotonin and cortisol have pro-insulin-resistance action. This means that an aggressive encounter, irrespective of the win–lose outcome, will be insulin sensitizing, but chronically shunning aggression will lead to a state of insulin resistance. The story does not stop at inducing insulin resistance. It also suggests a possible cause of β cell problems in the form of chronic deficiency of gastrin, EGF, and other growth factors needed for β cell regeneration. Further it accounts for deficiency of EGF, NGF, and BDNF which are likely to play an important role in diabetic neuropathy and retinopathy as we will see in a later chapter.
The positive feedback mechanisms stabilizing the diplomat state also explain why types 1 and 2 diabetes have very similar pathological consequences in spite of having very different etiologies. Aggression suppression is the cause of type 2 diabetes, whereas aggression suppression is an effect of type 1 diabetes. Insulin deficiency in type 1 diabetes results into disinvestment from muscle. This would induce aggression suppression through checkpoint mechanisms. The two types of diabetes have different entry points into the cycle, but the cycle runs in very similar way, and therefore, the downstream effects of both types of diabetes are highly overlapping if not identical.
After having tried to make a new synthesis, we need to address the question how we can differentially test the new theory in comparison with the old one. The answer is a little tricky but not very difficult to perceive. It is tricky because it is not a clear case of two hypotheses being completely different and nonoverlapping. It is not a simple case of differentiating between whether X causes A or Y causes A since Y causes X and X likely causes Y too. Fat and cholesterol modulate aggressive behavior, and aggression hormones affect fat metabolism. Therefore it is difficult to decide what comes first, what is really important, and what is not. But the subtleties of the suggested pathways and mechanisms help us in differentiating between the two. The aggression suppression hypothesis involves downregulation of testosterone, EGF, NGF, BDNF, and other growth factors in the suggested pathophysiological mechanisms. Therefore this hypothesis predicts altered levels of these markers preceding T2D and related disorders. If positive energy balance, obesity, and obesity-induced insulin resistance are the root cause of T2D, then there is no reason why one should find deficiencies of testosterone, EGF, NGF, BDNF, and other growth factors in diabetes. This therefore becomes a differential testable prediction. There is indeed reproducible evidence for deficiency of testosterone [86, 87, 104, 245, 336–338], EGF [152, 339], NGF [166, 340–344], and BDNF [165, 175, 345] in T2D. In addition there are a few more indications in support of the aggression suppression hypothesis:
1.
Anemia and iron metabolism: Aggression has a number of downstream physiological effects that must have evolved in anticipation of injuries. Just as salivary EGF is produced in anticipation of injuries, there are likely to be other preparatory alterations in anticipation of injuries. Injuries imply blood loss, and therefore, the body should be prepared for making up by increasing the rate of synthesis of hemoglobin and red blood cells. It is no surprise then that aggression stimulates erythropoietin synthesis through the agency of testosterone [346–348]. Erythropoietin is an important signal involved in red blood cell formation. Some diabetics become anemic to a variable extent. This anemia is generally not due to iron, vitamin B12, or any such dietary deficiency. It is erythropoietin deficiency that characterizes diabetes-associated anemia [349–354]. Anemia in diabetes is often associated with excess iron deposits in the liver, a condition called iron overload [355, 356]. It is even been suggested that iron overload may contribute to insulin resistance [355]. This indicates that the raw material for the synthesis of hemoglobin and red blood cells could be there, but the stimulus for synthesis is lacking which is very likely to be due to behavioral deficiency than dietary deficiency. The lipocentric paradigm does not have an adequate explanation for erythropoietin deficiency anemia and iron overload accompanying diabetes.
2.
Sexual dysfunction: Similarly the lipocentric view does not adequately account for sexual dysfunction accompanying diabetes. The aggression suppression hypothesis makes a stronger case supported by evidence. The aggression suppression mediator serotonin has been shown to decrease sexual motivation [357, 358]. Dopamine, on the other hand, has facilitative effect on sexual motivation [52]. Sex hormones of both sexes, testosterone and estrogens, are associated with aggression [359–361]. Dominant males that are sexually more active have higher testosterone levels than subordinate ones [362]. Obesity and metabolic syndrome, on the other hand, are associated with lower testosterone levels [86, 87, 104, 245, 336–338]. It is interesting to note that although erectile dysfunction in diabetic males has a positive correlation with plasma glucose levels [363–366], glycemic control does not improve erectile dysfunction [367], but chronic treatment of erectile dysfunction is insulin sensitizing [368]. This reflects on the possible direction of the arrow of causation.
3.
The origin of endothelial dysfunction: The three isoforms of NOS, namely, endothelial (eNOS), neuronal (nNOS), and inducible (iNOS), have complex effects on aggressive behavior as well as on metabolic syndrome. Male mice with targeted deletion of eNOS gene display dramatic reduction in aggression [217]. eNOS knockout mice were hypertensive and had fasting hyperinsulinemia, hyperlipidemia, and insulin resistance [218]. eNOS gene polymorphisms have a significant association with type 2 diabetes [219]. eNOS also has a significant role in facilitating wound repair and growth-factor-stimulated angiogenesis [369, 370]. Based on different model systems and different cell types/tissues examined, eNOS has both pro-inflammatory and anti-inflammatory effects [371], but it has a protective role against pancreatitis [302]. The effects of nNOS on aggression are sex specific in mice. In males nNOS knockout mice showed large increase in aggressive behavior and excess sexual behavior [220]. Inhibitors of nNOS also increase aggression in males [216, 221]. Female nNOS knockouts do not show increased aggression and may in fact show a decrease [222]. As opposed to eNOS knockouts which develop insulin resistance in both liver and peripheral tissue, nNOS knockouts develop only the latter [223]. It is apparent therefore that eNOS which has clearly a proaggression action is antidiabetic, and nNOS that has a mixed action on aggression also has mixed effects on insulin resistance. The inducible form iNOS has not been studied from the behavioral point of view, but targeted disruption of iNOS synthase protects against obesity-induced IR [224]. The role of NOS in sexual function is well known, and sildenafil, a popular drug used for the treatment of erectile dysfunction, has insulin-sensitizing effects [372], and interestingly, aggressive behavior has been noted as a “side effect” [373, 374]. Since nitric oxide signaling is directly involved in aggression, a chronic loss of aggression can affect NO levels which has important vascular effects. The traditional interpretation of diabetes holds raised plasma glucose levels to be responsible for vascular pathology. However signs of vascular pathology appear much before hyperglycemia. Therefore I feel that the traditional view has not adequately explained the origin of vascular pathology in diabetes.
There is one possible argument against the aggression suppression hypothesis. Aggression is characterized by sympathetic activation and suppression of aggression by increased parasympathetic tone. Contrary to our expectation, in T2D, there is increase in the markers of sympathetic tone. This contradiction is obvious but only apparent and stems from the naïve association of sympathetic activity with aggression. Although a physically aggressive act necessarily involves sympathetic activation, the reverse cannot be inferred. Sympathetic activation does not necessarily increase aggression. Evidence for association of sympathetic activation and aggression is contradictory [324, 375–379]. There are two reasons for the association being nonobligate. First is that the effect of sympathetic activation depends upon the standing metabolic, endocrinal, and behavioral state. For example sympathetic stimulation has a lipolytic action. But this action is mediated through β adrenergic receptors in visceral adipose tissue. The expression of β adrenergic receptors is testosterone dependent, and therefore, lipolytic effects of sympathetic action will be seen only if testosterone levels are adequate. Similarly sympathetic action will increase aggression only if all other factors are favorable for aggression. The other reason why sympathetic tone does not necessarily associate with aggression is that the effect of sympathetic activity on aggression is nonmonotonic. Moderate activation of central noradrenergic system increases aggression, whereas strong activation decreases it [376]. This makes sense for optimizing behavioral strategies. If sympathetic stimulation is induced by aggressive competition and the response is proportional to the level of threat, the nonmonotonicity can be easily explained. A weaker opponent poses less of a threat, and being aggressive is a fruitful strategy, but if the opponent is too strong the threat level is high, it is better to avoid aggression.
There are multiple reasons for which sympathetic activity does not have a one-to-one correlation with aggression. Therefore higher level of sympathetic markers in diabetes is not evidence against the aggression suppression hypothesis. There is a different reason for higher nonaggressive sympathetic activation in diabetes. This sympathetic activity is extremely important and central to diabetic hyperglycemia which we will see in Chap. 12. As yet we have not developed sufficient background to understand why sympathetic tone rises in diabetes. To understand this we need to wait a little more. But at this stage I want to make a subtle distinction. There is a fundamental difference in endocrine and neuronal signals in the body. Endocrine signals are like radio broadcasting. The signal reaches every corner more or less simultaneously and with the same intensity. Neuronal signals are specific to the recipient like a one-to-one telephone call. One muscle may be instructed to contract and the very neighboring muscle to relax at the same time. We should not forget that the autonomic system is a neuronal system primarily, although they also alter levels of epinephrine/norepinephrine generally. Owing to the specificity, one autonomic function could be on and while others are kept off. Therefore equating aggression with sympathetic activation and expecting that, since sympathetic tone is high in diabetes, aggression would aggravate diabetes is illogical.
Exercise and Aggression
Both the lipocentric and aggression centric views converge on the importance of physical activity or exercise. However there are important differences. Exercise is viewed in terms of burning calories by the lipocentric view, whereas exercise is a necessary component of physical aggression from the new viewpoint. There are two possible ways in which we can resolve between the two alternative ways in which exercise and its effects can be interpreted. First if exercise is only calorie burning, then all types of exercises with equal calorie expenditure should have equal effects on the endobolic state. The alternative view would expect a difference between the effects of different types of exercises, particularly between more aggressive and less aggressive exercises. By our definition, aggressive exercises comprise neuromotor actions involved in Stone Age hunting or fighting as defined in Chap. 5. So far there is little research investigating the effects of aggressive exercise, but experiments have shown that different types of exercises have different effects. For example, some studies found resistance exercise more insulin sensitizing than aerobic exercise [380–382]. Of particular interest is the finding that extremely short-duration (30 s) high-intensity exercises improve insulin sensitivity and other health parameters surprisingly well in spite of very low total calorie expenditure [383–386]. These exercises are similar to a short-duration intensive chase during hunting. Such exercises have been shown to increase aggression hormones and growth factors including testosterone, GH, and IGF binding protein [387]. Therefore it can be suspected that the beneficial effects of these exercises are through their aggression component rather than their energetic component. A yet unpublished study compared three groups of young males doing no exercise, less aggressive exercises such as swimming and running, and more aggressive exercises such as boxing or kickboxing. There was no difference in the insulin sensitivity and LDL cholesterol of the no exercise and nonaggressive exercise group, but the aggressive exercise group was significantly more insulin sensitive and had lower LDL and higher HDL levels [388]. More experiments specifically designed to test the effects of aggressive exercises in comparison with nonaggressive exercises are needed to resolve between the two alternative possible effects of exercise.
The other possible means of resolving between the two alternative interpretations is to look at the pathway through which exercise brings about its beneficial effects. If the lipid view is correct, the beneficial effects should be routed through energy pathways such as fat oxidation, glycogenolysis, and glycolysis pathways. If the aggression hypothesis is true, the effects should be brought about through pathways involving one or more of the aggression signal pathways described above including EGF, NGF, BDNF, endorphin, and sex hormones. Certain types of exercises upregulate testosterone [387, 389], EGF [390, 391], NGF [392, 393], and BDNF [394, 395] as well as endorphin [307, 308]. If exercise was related only to calorie burning, there is no reason why exercise should stimulate these molecules.
Mitochondrial function is said to be an important determinant of insulin resistance [396, 397]. If altered mitochondrial function has a causal role in insulin resistance, then it would be enlightening to look at the effects of lipids and those of exercise on mitochondrial function. Exercise training is reproducibly shown to improve mitochondrial function [398–401]. In synergy with exercise, the sex and aggression hormones testosterone and estradiol also enhance muscle mitochondrial capacity [336, 402], whereas corticosteroids reduce it [403]. On the other hand evidence for lipids or FFAs lowering mitochondrial function is shaky. In one study lipid accumulation actually increased mitochondrial proteins and their function [404]. If lipids were responsible for reduced mitochondrial function, lowering of lipids should have increased it. However, pharmacological lowering of plasma lipids in insulin-resistant subjects reduced mitochondrial gene expression instead of increasing it [405]. This strengthens the hypothesis that physical aggression and activity have direct insulin-sensitizing effects independent of their effects on lipid metabolism.
For an aggressive and routinely exercising individual, muscle building is of prime importance. Muscle building does not take place during exercise but takes place during postfeeding and resting time. For this to happen, muscle uptake of nutrients needs to be facilitated, and this is possible if muscles are insulin sensitive. Therefore there needs to be a mechanism that makes muscle tissue insulin sensitive upon exercising regularly. A new pathway with a large potential importance has come into light recently but is not yet completely worked out. It involves postfeeding hepatic production of a soluble signal molecule called HISS (hepatic insulin-sensitizing substance) that increases insulin activity specifically in muscle and thereby facilitates muscle building. HISS activity is shown to increase by exercise [406]. The compound responsible for HISS signaling is yet to be isolated, but available evidence shows that this pathway involves parasympathetic activity, NO, and insulin action in the liver [407–410]. It appears therefore that specialized mechanisms exist to fine-tune insulin sensitivity of muscle, and muscle insulin resistance is not a passive and inevitable process induced by fat. Exercise creates the need to invest energy in muscle, and the need stimulates the HISS pathway and perhaps other mechanisms to increase muscle insulin sensitivity independent of fat metabolism.
In reality the two effects of exercise, i.e., fat reduction and triggering aggression pathways, are not mutually exclusive, and both would be at work simultaneously. Research should then address the question of evaluating the relative importance of calorie burning versus aggression pathways in the beneficial effects of exercise. The increment in the levels of the above factors can be used to quantify the aggression component of an exercise schedule, and studies can be designed to evaluate the role of aggression component of exercise vis-a-vis the energy component. There is no research input in this question so far because the aggression component of exercise was not yet appreciated. This is evident by the fact that the otherwise comprehensive accounts of exercise physiology do not even mention that exercise stimulates EGF, NGF, BDNF, sex hormones, and other factors [411] although there is adequate evidence for it. Exercise physiologists need to consider the possibility that at least part of the exercise effect is mediated by the aggression pathways rather than the energy pathways.
Since evidence as well as pathways exists through which loss of aggression can lead to metabolic syndrome disorders, there is a need to explore the possibility further to test whether loss of aggression is both necessary and sufficient to cause any or all of the pathophysiological processes of this set of disorders. We should not repeat the mistake that was done with the classical theory of T2D where nobody really bothered to test whether obesity, insulin resistance, and insulin insufficiency comply to the necessary and sufficient criteria of causation for type 2 diabetes and all its complications. Since there is sufficient preliminary evidence for aggression deficiency to be the potential cause of insulin resistance or T2D, research in the near future should directly address the question whether it fulfills the necessary and sufficient condition for causation of insulin resistance and/or type 2 diabetes. Such experiments need to be sufficiently long drawn since the effects of behavioral deficiency are necessarily chronic. A note of caution is that the aggression that we address here is a normal and natural form of aggression innate to an individual’s physiology. There can be pathological forms of aggression that need to be carefully excluded. A pathological form of aggression is one that evades the physiological controls described in this chapter. For example, if the mechanisms of aggression checkpoint are impaired, a person with weak muscle or impaired hemodynamic response might still behave aggressively. This may or may not give the correlates of aggression that are expected in normal physiology. Such conditions are rare and will not disturb any study with a sufficiently large sample size and randomization. This warning is only for those who may like to come up with some isolated exceptional case and a pretense of having disproved me.
Some of the possible lines of investigations could be (1) to see whether there are strong, muscular, and physically aggressive individuals that are insulin resistant or type 2 diabetic. This needs to be studied in comparison with nonobese type 2 diabetics. We know that a significant proportion of diabetics belong to the latter. But there are no epidemiological studies looking at the former. Anecdotally I have seen many thin T2D patients, but I am yet to see one who is strong, muscular, and physically aggressive. Beyond personal impressions this question needs to be seriously addressed. If there is significant proportion of strong muscular and aggressive diabetics, it can be concluded that loss of aggression syndrome is not necessary for being a type 2 diabetic (just as being obese is not necessary). (2) If aggressive exercises in the absence of weight loss are unable to increase insulin sensitivity and other parameters of exercise benefit, it can be concluded that loss of aggression is not necessary for insulin resistance. Such experiments can be carefully designed by giving an intensive aggressive exercise regime along with sufficient caloric supplementation to ensure no loss of weight or loss of fat mass. (3) In the above experiment levels of testosterone, EGF, NGF, BDNF, and other growth factors can be monitored to test whether their levels increase in the absence of loss of weight or fat mass. (4) On the other hand, if a group of young and aggressive individuals is made to retract from physical activity and aggression for a sufficient duration and their weight is maintained at basal level by providing calculated dietary energy, if they fail to decrease their insulin sensitivity, loss of activity and aggression is not sufficient to induce insulin resistance. Here again the aggression markers can help in quantifying the loss of aggression. These and perhaps a few other ways would exist that can test the necessary and sufficient conditions for a causal relationship between loss of aggression and insulin resistance. It would not be easy to begin such experiments. Much thinking and pilot experiments would be needed to standardize and choose parameters. That would be the appropriate way to handle and develop the new way of thinking. This line of evidence will take a long time to come in hand but will prove to be the right direction of research.
There is a quick alternative way to test necessary and sufficient conditions for a putative causative factor of a phenomenon, that is, to construct an inclusive mechanistic model of the system as far as we know it. In such an artificial system perturbations can be introduced and the effect monitored. In the age of computers a simulated system can be a useful and powerful tool to study the possible effects of any change. This is like shaking a spider web at one place and seeing what moves and how. For this to work, the individual threads of the spider web need to be known based on which web can be constructed in a hypothetical cyberspace and then studied. Individual threads in this condition are single regulation reactions such as glucose stimulates insulin production, glucagon facilitates gluconeogenesis, or EGF stimulates β cell regeneration. With the involvement of some students and colleagues, we constructed a network model of all known signaling and regulatory pathways of the body concerned with T2D. The multiorgan physiological network consists of over 70 different players and more than 250 positive or negative regulatory actions. Only those regulatory actions were included that were clearly demonstrated in literature. We made an attempt to include all pathways that we could find. An attempt was made to exclude redundancy to keep the model to a simplest possible level. Even after that, it makes a very complex network (see Appendix II for details). Since most of the available information on signaling mechanisms is qualitative in nature, we look at the model qualitatively. Each node has only three possible states, normal, upregulated, and downregulated. An upregulated node can alter other nodes to which it is connected by positive or negative links. Through such links, a signal can travel through the entire network and change the state of the network.
Once such a network is constructed, one can introduce environmental perturbations singly or in combination and see the effect on the entire network. The network model can also be used to see the possible system-wide effects of any drug or remedial measure. In a complex network a large number of possibilities exist and analyzing such a network considering all possible perturbations and remedies is a huge amount of work. The analysis is far from being complete as of today. The network itself is perhaps incomplete since all regulatory pathways may not have been discovered so far. But even with an incomplete network, a network approach is certainly more comprehensive than reductionist thinking.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


