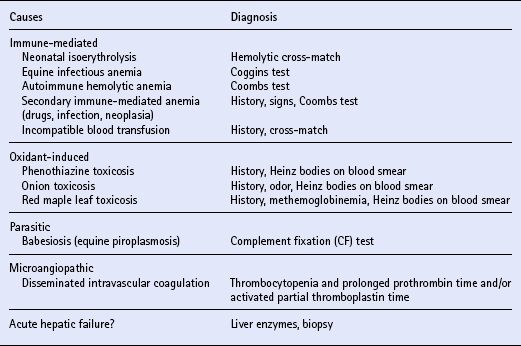
Chapter 9 During postnatal life, hematopoiesis occurs in the bone marrow. Maturation is attended by recession of hematopoiesis from the shafts of the long bones and the replacement of red marrow by resting yellow marrow; however, active hematopoiesis continues throughout life in the epiphyses of long bones and in the flat bones of the skull, vertebrae, sternum, ribs and pelvis. Transition from yellow to red marrow can occur in response to increasing demand for erythrocytes via the glycoprotein hormone erythropoietin (q.v.). The spleen functions only briefly in hematopoiesis during fetal life, but retains this potential into adult life. The unique architecture of the spleen and its fixed phagocytic cells bestow the important function of filtering from the blood aged or damaged cells, particulate debris and microorganisms. Hemoglobin from senescent erythrocytes is degraded and iron is stored within phagocytes until it is subsequently released into the plasma for reutilization during erythropoiesis. Splenic macrophages also perform a “pitting function” by removing inclusions such as Heinz bodies (denatured hemoglobin) or intra-erythrocyte parasites from erythrocytes. In the extrinsic pathway, factor Xa is formed via the action of tissue factor (TF), which accesses the circulation via inflammation or tissue necrosis. Fibroblasts in the blood vessel adventitia express surface TF constitutively, while vascular endothelial cells, tissue macrophages and blood monocytes are activated to produce TF by agonists such as endotoxin (q.v.). Anemia develops due to one or more of three pathophysiologic mechanisms: Equine erythrocytes are small (5–6 μm) and tend to adhere to each other to form a “stack” like coins (rouleaux). Marked rouleaux may be confused with autoagglutination, and this tendency for natural aggregation leads to rapid erythrocyte sedimentation from plasma. Equine plasma is normally quite yellow compared with that of other animals, due to the combined effects of blood carotenoids from green feed and the greater concentration of bilirubin. Fasting causes a marked increase in equine serum bilirubin and can result in clinical icterus (q.v.). There is no definitive explanation for fasting-induced hyperbilirubinemia in horses, but a net decrease in and/or competition for hepatic binding proteins (particularly ligandin) have been proposed. In consideration of hemolytic disease as a cause for icterus in horses, the influence of fasting must be determined. The clinical and laboratory findings of blood loss are largely determined by whether this occurs acutely, chronically, externally or internally. Com-mon causes for acute blood loss in horses include trauma to the limbs, post-castration hemorrhage, rupture of a uterine artery at parturition or erosion of the carotid artery by guttural pouch mycosis (q.v.). Hypovolemic shock (q.v.), characterized by tachycardia, tachypnea, hypothermia, pale and dry mucous membranes, prolonged capillary refill time, cold extremities and muscle weakness, generally develops when blood volume is reduced by more than 30%. Compensatory mechanisms are triggered immediately in an attempt to maintain circulating blood volume. Causes of chronic gastrointestinal blood loss include: parasitism (particularly large strongylosis); gastric or duodenal ulcers; non-steroidal anti-inflammatory drug (NSAID) toxicosis; and neoplasia (particularly gastric squamous cell carcinoma) (q.v.). Melena is rare. Because chemical tests for fecal occult blood are not highly specific, diagnosis of chronic gastrointestinal blood loss should be supported by a high index of clinical suspicion and ruling out other sources of hemorrhage. There are numerous causes and mechanisms for hemolytic anemia ( Table 9.1), which is regenerative anemia without hypoproteinemia. Intensified erythropoiesis is often associated with neutrophilia and regenerative left shift. Total and indirect bilirubin concentrations may be elevated. Other laboratory findings are determined by the cause of the anemia ( Table 9.1). In addition to a CBC, TPP and serum bilirubin, the diagnostic evaluation of suspected hemolytic anemia should include thorough blood smear examination, urinalysis, Coombs test and Coggins test (q.v.). Table 9.1 Reproduced with permission from Morris, D.D. (1989) Review of anemia in horses, Part II: Pathophysiologic mechanisms, specific diseases and treatment, Equine Practice 11: 34–46. Foals with NI are born clinically normal but subsequently develop depression, weakness and a reduced suckle response at 12–72 h. The rate and severity of disease are determined by the quantity and activity of absorbed alloantibodies. Affected foals have tachycardia, tachypnea and dyspnea. The oral mucosae are initially pale, then, in foals that survive 24–48 h, become icteric. Hemoglobinuria is rare. Cerebral hypoxia may induce seizures as a preterminal event. Laboratory findings include anemia and hyperbilirubinemia. Metabolic acidosis and azotemia eventually develop due to tissue hypoxia and the nephrotoxic effects of hemoglobin. The prognosis for NI in foals depends on the quantity and activity of absorbed antibodies and is indirectly proportional to the rate of onset of signs. Like most diseases, NI is much more effectively prevented than treated. Foals from any mare that previously produced a foal with NI should be provided with an alternate colostral source unless the sire has known blood type compatibility with the dam. Mares at risk of producing affected foals (negative for Aa and Qa alloantigens) may be identified by blood typing (q.v.). Stallions negative for Aa/Qa and suitable on the basis of other criteria may be difficult to identify. It is most reasonable to breed “at-risk” mares as desired then to screen their serum in the last month of pregnancy for the presence of alloantibodies. If alloantibodies (other than those to Ca) are detected, dam colostrum should be withheld and the foal provided with an alternative colostral source. The JFA test has been used to determine when it is safe for the foal to nurse. Definitive diagnosis of AIHA is based upon demonstration of patient antibodies that react with their erythrocytes. Autoagglutination is diagnostic of AIHA provided false agglutination is excluded. If true autoagglutination is not evident, diagnosis is most accurately made by agglutination in a direct antiglobulin (Coombs) test (q.v.), performed by incubating washed patient erythrocytes with appropriate dilutions of antiserum to equine IgG, IgM and complement components. A false negative Coombs test may occur immediately following a hemolytic crisis, or when corticosteroid therapy has been initiated. The osmotic fragility test should not be used for definitive diagnosis of AIHA, since positive results occur during other diseases that compromise erythrocyte membrane function (e.g. oxidative insult). Equine infectious anemia (EIA) (q.v.) is a multisystemic retroviral disease of Equidae, characterized by immune-mediated hemolytic anemia. Horses of all types are affected, but the disease is most prevalent in the southeastern USA. Because of its importance, EIA will be briefly considered separately from other causes of AIHA. Signs of subacute-chronic EIA result from virus-induced immunologically mediated tissue damage. Immune complex attachment to erythrocytes via the viral hemagglutinin produces hemolysis. Resultant anemia is worsened by decreased bone marrow erythropoiesis. Periodic disease flare-ups are due to the immune response to viral antigenic drift.
The hemolymphatic system
INTRODUCTION
HEMATOPOIESIS
HEMOSTASIS
Coagulation
ANEMIA
LABORATORY EVALUATION
BLOOD LOSS ANEMIA
HEMOLYTIC ANEMIA
IMMUNE-MEDIATED HEMOLYTIC ANEMIA
Neonatal isoerythrolysis
Clinical signs and laboratory findings
Prognosis and prevention
Autoimmune hemolytic anemia
Diagnosis
Equine infectious anemia
Etiology and pathogenesis
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The hemolymphatic system
Only gold members can continue reading. Log In or Register to continue
