Fig. 6.1
Epigenetic mechanisms. DNA methylation, histone modifications and non-coding RNAs (nc-RNA) are often associated with changes in transcriptional activity
The best-known epigenetic mark is DNA methylation (Esteller 2008). This is a dynamic process that takes place throughout the course of development in multi-cellular organisms and ensures the maintenance of normal expression patterns. DNA methylation is involved in many processes including genomic imprinting (Feinberg et al. 2002), the gene-dosage reduction involved in X-chromosome inactivation in females (Payer and Lee 2008), and silencing parasitic and foreign elements (Doerfler 1991), among other processes.
DNA methylation is carried out by a family of enzymes called DNA methyltransferases (DNMTs), which transfer a methyl group from the donor S-adenosyl-methionine (SAM) to the DNA base. In mammals, this family of enzymes includes DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3-like (DNMT3L) (Bestor 2000), each of which has a specific function although only DNMT1, DNMT3A, and DNMT3B have demonstrated DNA methyltransferase activity (Okano and Li 2002). DNMT1 is considered the principal enzyme responsible for the maintenance of DNA methylation patterns following cellular replication (Fuks 2005), while DNMT3A and DNMT3B have been linked to de novo methylation during embryonic development (Okano et al. 1999; Chen et al. 2003). DNA methylation occurs in cytosines that precede guanines, which are called CpG dinucleotides. CpGs are not randomly distributed in the human genome, rather there are CpG-rich regions, known as CpG islands, which span the 5′ end of the regulatory region of many genes. Whilst these islands are usually non-methylated in normal cells, DNA methylation alterations related to different human pathologies, developmental processes and aging have been found (Urdinguio et al. 2009; Fernandez et al. 2012). In particular, DNA methylation alterations have been widely studied in cancer, where it has been found that specific hypermethylation in the CpG islands of tumor suppressor genes causes their inactivation and, the consequent loss of their protective function in tumor development (Jones and Baylin 2002; Esteller 2008; Fernandez et al. 2012). DNA methylation has been studied extensively in relation to reproductive biology, where not only has it been characterized in germ cells and during the different stages of development, but it has also been related to various disorders, such as those linked to imprinting (Sandhu 2010).
Apart from DNA methylation, other important epigenetic mechanisms are involved in chromatin regulation, including gene expression control resulting from the reversible modification of the amino-terminal tail of histones (Fraga and Esteller 2005). There are in fact at least eight types of posttranslational modifications found on histones, of which lysine and arginine acetylation, lysine methylation, serine phosphorylation and lysine ubiquitination are the most thoroughly studied. The combination of the different histone modifications constitutes the histone code (Jenuwein and Allis 2001). This code determines chromatin function in the nucleus context regarding, for instance, the chromatin packaging state and, therefore, gene activity in specific chromatin regions. Thus, different histone modifications have been associated with activation or repression of transcription. The most extensively researched histone modification is, in fact, acetylation, which is generally associated with active gene transcription (Allfrey et al. 1964), whereas methylation is linked to either activation (i.e., H3K4, H3K36, and H3K79) or repression (i.e., H3K9, H3K27, and H4K20), depending on the conditions or residue modified (Kouzarides 2007). These modifications are mediated by histone acetyl transferases (HATs), histone methyl transferases (HMTs), histone deacetylases (HDACs), and histone demethylases (HDMs), among other enzymes. The characterization of histone modifications in relation to reproduction and the different stages of development is on-going although it has been associated with the processes of gametogenesis and embryogenesis (Sasaki and Matsui 2008).
In recent years the non-protein-coding portion of the genome has been shown to be crucial for normal development and function as well as for disease (Mercer et al. 2009; Esteller 2011). Its functional relevance is especially noticeable in the case of a class of small non-coding RNAs (ncRNAs) called microRNAs (miRNAs) (He and Hannon 2004; Mendell 2005). Both epigenetic and genetic defects in miRNAs, as well as their processing machinery, have been found in human diseases, particularly cancer (Esquela-Kerscher and Slack 2006; Hammond 2007; Croce 2009; Nicoloso et al. 2009). However, miRNAs are just a small element of a complex and as yet poorly understood picture, and other ncRNAs might also play a role in the development of many different disorders (Mercer et al. 2009; Esteller 2011). ncRNAs consist of various RNA species that are not translated and evolutionarily conserved among organisms. One single ncRNA may control hundreds of genes. In complex organisms, the developmental and tissue specific regulation of many genomic sequences (Carninci et al. 2005; Kapranov et al. 2007) has promoted the characterization of the different types of ncRNAs that are transcribed in human cells. Based on their length, ncRNAs can be divided into short ncRNAs including small interfering RNAs, miRNAs and PIWI-interacting RNAs (piRNAs); intermediate ncRNAs like small nucleolar RNAs (snoRNAs); and the heterogeneous group of lncRNAs (including large intergenic non-coding RNAs (lincRNAs) and transcribed ultraconserved regions (T-UCRs) among others). Thus far, most of the work has focused on short RNAs, such as miRNAs, however long non-coding RNAs (lncRNAs) (Kapranov et al. 2007), are recently attracting attention.
In terms of reproductive biology, ncRNAs are not as well-studied as DNA methylation or histone PTM. However, several short ncRNAs have been related to germ cell development (Banisch et al. 2012), specially miRNAs have been demonstrated to participate in the physiology and development of gonadal cells in mammalian reproduction (Hossain et al. 2012). Recently, the role of lncRNAs in this process has also begun to be revealed (Sendler et al. 2013).
In this chapter, it will be explained the epigenetic mechanisms that are related to reproductive aspects and how environmental factors, such as exposure to toxic substances or treatment with assisted reproductive techniques (ART), may be involved in epigenetic alterations that affect the reproduction process.
2 Environmental Epigenetics
Epigenomes change during ontogenic development and aging. Some of these changes have natural biological functions, such as defining specific developmental stages (Reik 2007; Feil and Fraga 2012), In addition to natural epigenetic changes associated with development, there are also apparently random variations with, seemingly, no biological function. These random epigenetic changes may be mediated by both intrinsic and extrinsic factors (environment), and are considered to be epigenetic alterations (“epimutations”) (Feil and Fraga 2012).
The term “environmental epigenetics” refers to the interaction between the environment and the epigenome, which is susceptible to undergoing modifications. Although how environmental factors can cause negative epigenetic changes remains largely unknown, alterations in DNA methylation or histone modification patterns may induce changes in normal gene expression, which in turn could be associated with a wide range of diseases including various reproductive disorders (Cortessis et al. 2012). Given the absence of mechanisms to repair epimutations, at least none that are known, it is to be expected that the effect of disturbances caused by the environment are greater in the epigenome than in the genome (McCarrey 2012).
Modifications can occur in the epigenome throughout life, but to better understand the impact the environment has it is necessary to consider two different scenarios: embryonic development and adult life (Aguilera et al. 2010). The most vulnerable period is during embryogenesis due to high level of cell division, and the fact that it is when epigenetic marks are undergoing critical modifications (Dolinoy et al. 2006). Moreover epigenetic alterations in this period can be transmitted over consecutive mitotic divisions and affect more cells than those occurring in adults during postnatal development. The placenta is especially important during fetal development, and its functions can be altered or influenced by the environment which may result in pregnancy problems such as early pregnancy loss, preterm birth, intrauterine growth restriction (IUGR), congenital syndromes, and preeclampsia, which have all been linked to epigenetic alterations (Robins et al. 2011).
There is some evidence that epigenetic alterations underlie the associations found between adverse environmental conditions during early developmental stages and later adult disease. Developmental Origins of Health and Disease (DoHAD) is a hypothesis based on the concept of “developmental plasticity” (Hales and Barker 2001), which could explain how environmental exposures during developmental periods can cause alterations which may increase the risk of disease and dysfunction later in life (Barouki et al. 2012). As such, the mother’s lifestyle—diet, obesity and alcohol—and tobacco consumption—during pregnancy or the characteristics of the placenta—size of the uterus, availability of nutrients could affect the epigenome of the offspring.
The DoHAD hypothesis is supported by evidence that dietary restrictions during early development in mammals have been associated with the onset of various diseases during lifetime, including cardiovascular or metabolic disorders, or even cancer (Perera and Herbstman 2011).
Different epigenetic mechanisms may be mediating the effect of such nutritional conditions on the appearance of altered phenotypes. A very good example of these associations comes from studies of the offspring of women pregnant during the Dutch Hunger Winter (1944–1945) (a famine that took place in the Netherlands during World War II). Famine exposure in the peri-conceptional period, which is particularly sensitive to changes in the diet of the mother, led to adverse metabolic phenotypes and mental phenotypes in the next generation (Heijmans et al. 2008). Individuals prenatally exposed to famine during this period presented less DNA methylation of the imprinted insulin-like growth factor 2 (IGF2) gene compared with their unexposed siblings (Heijmans et al. 2008). Interestingly this gene may play a role in the development of certain diseases, including coronary heart diseases, one of the diseases that has been found associated with food deprivation during the gestational period. Another classic example that illustrates the effect of diet on the phenotype during the period of gestation comes from the Agouti viable yellow (A vy ) mice model (Cropley et al. 2006). The Agouti gene is responsible for determining whether a mouse’s coat is banded (agouti) or of a solid color (non-agouti), and is regulated by the DNA methylation status of an intra-cisteral A particle (IAP) inserted upstream of the canonical wild-type transcription start site. Methyl-donor supplementation during gestation has been demonstrated to affect the epigenetic status of fetal germ cells and, accordingly, the coat color of the offspring. These epigenetic changes induced by maternal diet were maintained in gametogenesis and embryogenesis of the progeny. Furthermore, the mentioned diet supplementation was shown to affect not only the F1 but also the F2 (Cropley et al. 2006).
Due to their adverse effects on reproduction it is important to emphasize the importance of exposure to endocrine disruptors during pregnancy. Endocrine disruptors are chemicals that at certain doses can interfere with the endocrine (or hormone) system in mammals. They can be classified by their chemical composition into: pesticides (DDT and methoxychlor), fungicides (vinclozolin), herbicides (atrazine), industrial chemicals (PCBs, dioxins), plastics (phthalates, bisphenol A (BPA), alkylphenols) and plant hormones (phytoestrogens) (Skinner et al. 2011). Apart from these chemical products, some pharmaceuticals, personal care products and nutriceuticals are also known to be endocrine disruptors (Daughton and Ternes 1999). The majority of endocrine disruptors are not actually able to alter DNA sequence, but their most significant long term action appears to be related with alterations in the epigenome where they can affect normal reproductive physiological development and functions by acting as weak estrogenic, antiestrogenic, or antiandrogenic compounds. According to the DoHAD hypothesis, abnormal actions of endocrine disruptors during pregnancy can have drastic effects with regards diseases in later life. Females exposed to an excess of androgens early in gestation exhibit increased susceptibility to diseases such as polycystic ovaries in adult life (Abbott et al. 2005). In adult males, perinatal or pubertal exposure to compounds such as estradiol and BPA alters the prostate epigenome and heightens susceptibility to carcinogenesis in adult males (Prins et al. 2008). The mechanisms ways in which such endocrine disruptor exposure in early life is able to promote an adult onset effect in an organ are assumed to involve, at least in part, epigenetic mechanisms.
Besides endocrine disruptors, other pollutants can also alter epigenetic patterns during the prenatal stages: For example, in humans, arsenic exposure in uterus has been associated with increased lung cancer in adulthood. Exposure to tobacco smoke has also been associated with several diseases such as respiratory and metabolic diseases or cancer in children exposed in the womb, which in turn showed abnormalities of methylation patterns (Perera and Herbstman 2011). In addition, exposure to atmospheric pollutants like polycyclic aromatic hydrocarbons (PAHs) can also have adverse effects on fetal growth and have been associated with a decrease in global DNA methylation in cord blood cells of newborns exposed in the womb (Perera and Herbstman 2011).
Although the epigenome of differentiated cells is relatively stable, it too can be altered by environmental conditions during the postnatal stages. It is important to take into account that epigenetic patterns are tissue or cell specific and that not all tissues are equally exposed, hence the effects of environmental factors on the epigenome of an organism may depend on tissue type. The effects of environmental factors during adulthood may also depend on lifestyle conditions including diet, living place and/or workplace, pharmacological treatments, and unhealthy habits (Aguilera et al. 2010). For example, many dietary components have been shown to be linked to DNA methylation changes, and others to have the capacity to influence the activity of HDACs (Feil and Fraga 2012). Useful examples of how the environment affects the epigenome come from monozygotic twin (same genotype) studies, showing that in addition to the increase in epigenetic differences between identical twins over time, different lifestyles may contribute to heighten these differences, and even explain the differential appearance of diseases (Fraga et al. 2005). These epigenetic changes are small and most probably cumulative and develop over the lifetime of the individual, making it difficult to establish the relationship between environmental factors and epigenetic changes (Baccarelli and Bollati 2009).
Apart from nutrition, there are other environmental exposures such as to air pollutants, metals, tobacco smoke, drugs, sun, or alcohol that can alter the epigenetic patterns in the adult stage (Belinsky et al. 2002; Bleich et al. 2006; Baccarelli and Bollati 2009; Christensen et al. 2009; Gronniger et al. 2010; Langevin et al. 2011). Epidemiological studies have determined that exposure to air pollutants may cause not only alterations in global DNA methylation in blood (Baccarelli and Bollati 2009; Baccarelli et al. 2009), but also specific DNA hypermethylation of tumor suppressor genes such as the tumor protein p53 (p53) and cyclin-dependent kinase inhibitor 2A (p16) in arsenic exposed subjects (Chanda et al. 2006). Exposure to the carcinogen benzene has also been associated with changes in DNA methylation, including a significant reduction in global methylation and hypermethylation of cyclin-dependent kinase inhibitor 2B (p15) and hypomethylation of the melanoma antigen family A, 1 (MAGE-1) (Bollati et al. 2007). Another environmental contaminant of note is the endocrine disruptor BPA, and as we have seen in relation to the prenatal stages, exposure to this compound may be associated with epigenetic alterations (Hanna et al. 2012). In addition, exposure to this product has been related to breast and prostate cancer, polycystic ovarian syndrome and male infertility, among other diseases (Markey et al. 2001; Ho et al. 2006; Kandaraki et al. 2011; Li et al. 2011).
One of the most important aspects of the effect of environment or environmental conditions on the epigenome is the impact that these can have on trans-generational inheritance, i.e. the transmission of epigenetic changes induced by the environment from one generation to another. At this point it is important to distinguish between trans-generational epigenetic effects and trans-generational epigenetic inheritance. The former include the effects of environmental exposures on adults, which is capable of altering the phenotype of their offspring via the placenta or breastmilk in mammals. The latter, which will be discussed in more detail in the following sections of this chapter, refers to transmission via the gametes. Although most epigenetic alterations that occur in germ cells are reversed during epigenetic reprogramming in gametogenesis, some are able to evade this control and are thus transmitted to the next generation.
3 Epigenetic Reprogramming in Germ Cells and Early Embryo Development
In mammals, genome-wide epigenetic reprogramming mainly occurs at two stages of development: during primordial germ cell (PGC) development and during the early stages of embryonic development following fertilization (Sasaki and Matsui 2008; Feng et al. 2010; Kota and Feil 2010) (Fig. 6.2). Environmental exposures during these two periods can consequently particularly affect the offspring. In both stages, DNA methylation patterns are erased and followed by re-methylation. Together with these DNA methylation changes, there are also rearrangements in the post-translational modifications (PTMs) of some histones. However, changes in DNA methylation and PTMs differ slightly depending on the gender of the germ cell that will be generated (Sasaki and Matsui 2008).
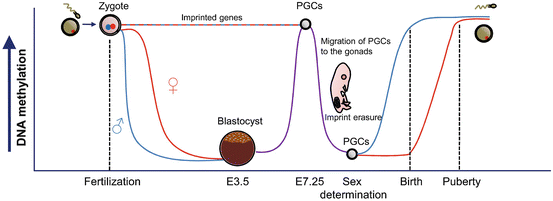
Fig. 6.2
DNA methylation changes during epigenetic reprogramming in mammalian germ cells and early embryo development. PGCs Primordial germ cells
An increased knowledge of the epigenetic mechanisms involved in those processes will help to understand, for instance, infertility-associated pathologies or failures occurring during the application of assisted reproductive technology (ART) in humans.
In mammals, PGCs originate in the epiblast during embryonic development. In mice, it has been shown that germ cells begin to suffer epigenetic changes such as histone H3K9 dimethylation (H3K9me2) loss or DNA methylation decrease from embryonic day 7.5 (E7.5) until approx. E11.5; changes which coincide with germ cells migration to the developing gonads (Fig. 6.2). Concurrently, an increase in H3K27me2 has been detected until E13.5 (Sasaki and Matsui 2008). When PGCs have settled in the gonads (E11.5), the erasure of parental imprints occurs and the inactive female X chromosome is thought to be reactivated. Although at this stage almost all DNA sequences have suffered demethylation, some sequences such as intra-cisteral A particle (IAP) and a few long terminal repeats (LTR) sequences avoid the complete loss of DNA methylation (Sasaki and Matsui 2008; Smith and Meissner 2013) indicating that some epigenetic marks related to these regions, and their possible alteration in response to environmental factors, could be heritable and transmitted to the next generation. A classic example of this heritability is the Agouti viable yellow (A vy ) mice mentioned in previous sections.
It is still unclear how the DNA demethylation process is regulated during this phase, given that it could be related to either passive or active demethylation mechanisms. The latter would involve DNA deaminases or the 5-methylcytosine (5mC) dioxygenases TET1 and TET2, which trigger, in response, the repair of pathways, especially base excision repair (BER) (Feng et al. 2010).
After the erasure of parental imprints, there is a re-establishment of DNA methylation patterns (Fig. 6.2), which are dependent on the gender of the generated germ cell. While paternal imprinting is established during early stages of spermatogenesis in the fetus from E14.5 until birth, maternal imprinting only occurs after birth during oocyte growth and finishes before maturation, during puberty (Sasaki and Matsui 2008; Smith and Meissner 2013). During this process not all sequences from both types of gametes re-methylate in the same way. While some repeats, such as LINE1, are more methylated in male than in female gametes, the latter show higher methylation levels in IAP sequences than male gametes. Although the mechanisms involved in these re-methylation processes are still unknown, it seems that de novo DNMTs and small non-coding RNA molecules could be involved (Sasaki and Matsui 2008; Smith and Meissner 2013). Defects in the re-methylation process of male germ cells in genes, such as the imprinted maternally expressed transcript H19 and mesoderm specific transcript MEST, may be associated with infertility (Rajender et al. 2011) and abnormal methylation of several imprinted genes in human sperm, with oligozoospermia (Marques et al. 2008; Kota and Feil 2010), indicating the importance of epigenetic mechanisms in the regulation of reproduction.
Apart from differential DNA re-methylation, post-translational modifications of histones also differ between male and female germ cells during meiosis and gamete maturation processes. Male germ cells suffer several changes in histone methylation and acetylation patterns during the pre-meiotic stage and the initial stages of Prophase I in the first meiotic division (Sasaki and Matsui 2008). Later, during male gamete maturation (spermiogenesis), as well as changes in some histone mark patterns, there is also a process of histone-protamine exchange, which induces spermatozoa chromatin compactness and seems to function to give the genome of the germ cell protection against environmental damage (Sasaki and Matsui 2008; Kota and Feil 2010). This process whereby histones are replaced by protamines is not complete, and 5–15 % of the histones are retained in mature sperm and play an important role in development (Hammoud et al. 2009). In contrast, changes in histone methylation and acetylation patterns in female germ cells occur from final the stages of Prophase I of the first meiotic division until oocyte maturation following puberty (Sasaki and Matsui 2008; Kota and Feil 2010).
The other important round of epigenetic reprogramming is produced in early embryo development, immediately after fertilization and zygote formation (Fig. 6.2). At this point, global DNA methylation loss (with the exception of imprinted genes) begins and lasts until the blastocyst stage (E3.5). Afterwards, DNA methylation gain occurs until approximately E7.5, when a new cycle of reprogramming starts (Perera and Herbstman 2011; McCarrey 2012; Smallwood and Kelsey 2012; Smith and Meissner 2013) (Fig. 6.2). Similar to the demethylation processes in PGCs, it seems that base excision repair (BER) is associated with DNA demethylation in the zygote (Kota and Feil 2010). Furthermore, like the epigenetic reprogramming in PGCs, these demethylation processes are coupled with changes in histone mark patterns although in this case, however, they are greater in the paternal than in the maternal genome (Seisenberger et al. 2013; Smith and Meissner 2013).
Given all the above, it seems reasonable to assume that these fine epigenetic reprogramming processes may be particularly susceptible to changes that might even compromise reproductive success. Although the mechanisms by which environmental factors may influence the success of this “epigenetic reset” are not clear, the most important point to note is that changes occurring in germ cell precursors or gametes can be transmitted to the next generation.
4 Epigenetic Alterations in Germ Cells
Although environmental factors can modify the epigenome of somatic cells, only when these changes occur in the germ cell line can they be trans-generationally transmitted (Skinner et al. 2011). The transfer of heritable material from parents to progeny through the germ line is known as “transgenerational” change and to be defined as such changes must continue at least through to the F3 generation (Anway et al. 2005). When an F0 is exposed to environmental factors during gestation, the F1 embryo is also exposed and changes in its germ line can result in the transmission of the alteration to a subsequent F2. However, when these changes are present in F3, further transmission occurs in the absence of direct exposure, and the resulting transgenerational phenotypes may be maintained for generations (Fig. 6.3). In germ line development, permanent alteration in epigenetic programming appears to be the mechanism involved in the transgenerational phenotype (Jirtle and Skinner 2007). Nonetheless further research in this area is required to determine how epigenetic alterations are transmitted down the generations.
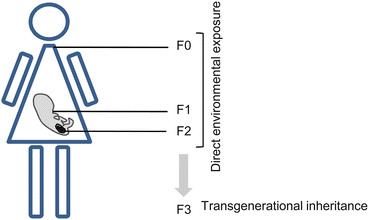
Fig. 6.3




Transgenerational epigenetic inheritance. Scheme showing how direct exposure to environmental factors during pregnancy can affect the epigenome of the F0 (mother), F1 (embryo), and F2 (germ-line). Trans-generational inheritance refers to the transfer of epigenetic changes to the F3 generation (without its direct exposure)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
