Fig. 1.
Exemplary recordings showing SD in a mouse brain slice after stepwise increase of [K+] in the bathing medium with (red) and without (blue) ouabain (5 μM). Recordings have been obtained employing K+ selective/reference electrodes located in layer II/III of the somatosensory cortex. The specific Na,K-ATPase inhibitor ouabain reduces the SD threshold and prolongs the accompanied extracellular K+ surge as well as the slow field potential shift.
Intracellular recordings from neurons or glial cells allow characterization of passive membrane properties, neuronal firing characteristics, currents, and synaptic responses on a single-cell level. Intracellular recordings in whole-cell configuration can be performed using either high-resistance sharp electrodes or patch electrodes. Resting and passive properties of neurons, such as resting membrane potential, input resistance, and membrane time constant can be determined. Moreover, cellular firing characteristics can be assessed, such as the threshold for action potential initiation and firing rate in response to current injection (current steps/ramps) or by synaptic activation through application of electrical stimulation of afferent fibers. In addition, intracellular recordings allow for measurement of excitatory and inhibitory postsynaptic potentials and currents (EPSP, EPSC, IPSP, and IPSC). For smaller currents in the range of 5–20 pA (e.g., miniature EPSPs/EPSCs), as well as for single-channel recordings, utilizing the patch-clamp technique is indispensable.
We apply whole-cell recordings in current-clamp or voltage-clamp mode using sharp pipettes and a discontinuous single-electrode voltage clamp amplifier (dSEVC, “switch-clamp” type) that balances out the resistive influence of the recording micropipette and allows single-electrode measurements. Sharp glass microelectrodes are filled with 2 M potassium acetate and yield impedances of 60–90 MΩ. Micropipettes are pulled from borosilicate glass (outer diameter 1.2 mm). Signals are amplified (SEC-10L, NPI Electronics, Tamm, Germany), low-pass filtered at 2 kHz, displayed on an oscilloscope, digitized at 10 kHz (CED-1401, Spike software, Cambridge Electronics Design, Cambridge, UK), and stored for off-line analysis. Acceptable cells should display stable resting potentials of at least 50 mV, total neuron input resistances of 35–80 MΩ, and overshooting action potentials. Action potentials are elicited either by synaptic stimulation or direct ramp depolarization (100–400 pA). The neocortical neuron population is identified on the basis of their electrophysiological properties (28) and by means of morphology through injection of Alexa biocytin and fluorescence microscopy (29). The electrode tip can be filled with various kinds of dyes, including Lucifer yellow or biocytin, for morphological studies.
To investigate the process of SD itself in neurons and neuroglial cells (30, 31), it has been suggested to use low-resistance patch electrodes because of the large increase in membrane conductance and the considerable swelling of the tissue (32). If intracellular measurements are to be performed during SD, an important issue is the correction of the measured intracellular potential (V i) for the large extracellular potential shift (ΔV m = ΔV i − ΔV o) (5, 32, 33).
4 Inducing Sustained Depolarization and Determining the Threshold
Spreading depolarization is a very robust response of the gray matter of the central nervous system that is readily triggered by various means, such as electrical stimulation, mechanical stimuli, alkaline pH, hyperosmolarity, and elevated concentrations of potassium, glutamate, and ouabain (34). In the brain slice preparation, we mostly employ protocols that make use of elevated [K+] as a trigger for SD—focally under normal aCSF or globally by increasing [K+] in the bathing solution ([K+]aCSF). We induce SD focally using a highly concentrated KCl solution (e.g., 1–3 M KCl) either by droplet application onto the surface of the brain slice or by microinjection into tissue by means of glass micropipettes with a resistance of about 200 kΩ. To determine SD threshold, pressure-ejection pulses of increasing duration (at 5–10-min intervals in 25-ms steps) can be applied utilizing a pressure ejection system (e.g., Ionophor 3 pressure unit, Science Products; PDES-02DX, npi Electronic or Picospritzer, General Valve). In order to determine SD threshold under extensive and prolonged exposure to elevated [K+], we either raise [K+]aCSF in steps of 2.5 mM at 30-min intervals or raise [KCl]aCSF to a fixed concentration (e.g., 22.5 mM) in the bathing medium ([Na+]aCSF is lowered accordingly to maintain iso-osmolality).
Parameters, such as threshold [K+]o, as measured with ion-selective/reference electrodes and time elapsed until SD is ignited can be determined. Alternatively, SD can be provoked by electrical stimulation through application of trains of voltage- or current-controlled square waves of 50–100-μs duration at a frequency of about 20–50 Hz up to maximal stimulation intensities over 3–10 s. Stimulation intensities can be adjusted to determine the threshold for SD induction. However, as far as our experience goes, this method is associated with a high failure rate to induce SD in untreated brain slices under normal conditions. The same applies to single-electrical pulses with longer duration (100–200 ms) and higher charges, which we found to increase the risk for tissue damage. Nevertheless, we have obtained good results employing electrical induction of SD in vivo, associated with a low failure rate and reproducible results. To induce SD electrically, we position a bipolar stimulation electrode onto the intact dura mater, exposed by preparing a small cranial window (Ø ∼ 4 mm). Then, biphasic pulses of 100-ms duration with increasing intensities up to 1 mA at 5-min intervals are applied until SD is triggered.
5 Optical Imaging of SD In Vitro
Fluorescence microscopy empowers investigators to assess functional aspects by measuring ion variations (e.g., Na+, K+, Cl−, Ca2+, Mg2+, and Zn2+) and pH and to detect changes in membrane potential of whole cells or mitochondria using voltage-sensitive fluorophores. In addition, insights regarding the metabolic state of the tissue can be gained from monitoring the autofluorescence of key components of oxidative phosphorylation, like nicotinamide adenine dinucleotide (NADH). The development of more advanced imaging techniques, in particular multiphoton microscopy, now permits 3D functional imaging in brain tissue, even in specific cell compartments (e.g., dendrites and spines), and transgenic technologies can create organisms that produce their own fluorescent chimeric molecules. In contrast to fluorescence microscopy, intrinsic optical signals (IOS) can be detected over a broad waveband of light and do not require loading of foreign substances into the tissue.
5.1 Intrinsic Optical Signals
Assessment of IOS is perhaps the most commonly used imaging approach to study SD in vitro and in vivo. IOS represent alterations of optical properties of unstained tissue which can be measured as a change of either light transmittance or reflectance. Measurement of IOS does not necessitate any tissue manipulation other than illumination of the preparation. The sustained signal changes feature comparably large amplitudes and can be assessed with a charge-coupled device (CCD) camera mounted to a microscope or an array of photodiodes. Detectable IOS in brain tissue can be induced by intense neuronal activity (e.g., induced by repetitive stimulation or during seizure-like activity), osmotic changes leading to swelling or shrinkage of neurons, and SD. In contrast to intense neuronal activation or mild hypotonicity and despite the swelling of brain cells with subsequent shrinkage of the extracellular space (35–38), the main optical change during SD is an increase in light scattering (39) that coincides with the extracellular DC potential shift. The decrease in light transmittance has been shown to be Cl− dependent (40) and has been hypothesized to be due to swelling of intracellular organelles (e.g., mitochondria) (41). Other findings indicate that it could be the consequence of structural changes, located mainly in dendritic processes (36, 42). The advancing wave of optical changes accompanying SD can be roughly resolved into three phases (43) starting with a brief, weak increase followed by a large decrease, succeeded by a weaker and prolonged increase of light transmittance. During the course of SD, recording IOS permits 2D mapping of the spreading event, thereby allowing the calculation of the propagation velocity and the extent of spread. If quantification of the IOS is intended, a scientific-grade CCD microscope camera, which usually features cooling for noise reduction and 12-bit signal resolution with a linear output, should be employed (44). The linear output feature refers to a direct linear relationship between the number of detected photons and the corresponding pixel values. Compared to assessing synaptic activation using IOS, SD is a slow process and low frame rates will suffice (e.g., 5 Hz or less) to monitor the temporospatial feature of the spread of SD. A stabilized light source, equipped with a xenon or halogen lamp, should be used to avoid fluctuations which would interfere with the analysis of the IOS. In general, image acquisition is followed by processing of the image data consisting of subtraction of the images which are then overlayed (pseudocolored) on the original image (Fig. 2). Regions of interest (ROIs) can be defined and the average light intensity within the ROI is determined over a time series of images. In our laboratory, optical changes are monitored by transilluminating the slices from below using a halogen lamp and a stabilized voltage-regulated power supply (KL 1500, Schott). Homogenous illumination is achieved by an arched glass rod built into the interface chamber that conducts the light to the transparent floor of the chamber. Light transmission changes are recorded using a microscope-mounted camera (DFC 360FX, Leica) that yields 12-bit bitmap image files which are acquired at a frame rate of 5 Hz, digitized, and stored for further processing. The first frame (T 0) in a series, captured before the onset of SD, serves as control and is subtracted from subsequent images of the series (∆T = T x − T 0) and divided by the control image to obtain the relative light transmission change:
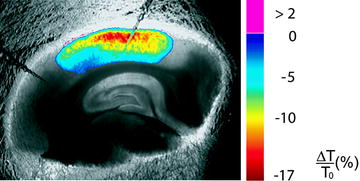
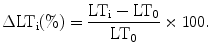
Fig. 2.
Imaging of SD using IOS in the acute mouse brain slice. Pseudocolor overlay represents relative changes in light transmission. Two ion-selective/reference electrodes are placed in a midcortical layer.
ROIs are placed around the recording electrodes to quantify LT changes. SD velocity is then determined by the propagation of the transient LT change.
5.2 Two-Photon Laser Scanning Microscopy
Modern imaging techniques, such as two-photon laser scanning microscopy (2PLSM) (45), have greatly contributed to our understanding of cellular physiology and dynamics of many cell types in brain slices in vitro as well as in in vivo preparations. Imaging with 2PLSM is also useful for high-resolution cellular monitoring during SD. With 2PLSM, problems associated with scattering and absorption of photons by neural tissue are significantly reduced as compared with one-photon excitation used in confocal and wide-field fluorescence microscopy (46). In 2PLSM, the use of the long-wavelength excitation photons results in less scattering and low absorption by water and intrinsic tissue chromophores and provides better tissue penetration. Simultaneous absorption of two photons that combine their energies excites a chromophore and generates fluorescence that can be acquired from several hundred microns deep within living tissue. The intensity-squared requirement of two-photon excitation restricts chromophore excitation and emission to a narrow focal plane (focal slice), where the laser beam is focused. This reduces photobleaching and photodamage in thick living tissue, permitting repeated sampling (47–49). The intrinsic confocality of two-photon excitation results in thin optical sections that can be stacked to create a 3D image.
In recent years, generation and availability of transgenic mice expressing fluorescent proteins in a small percentage of brain cells have greatly facilitated imaging by providing the opportunity to capture detailed time-lapse images to observe the dynamic properties of living cells. Here, we summarize how we have used 2PLSM in live cerebral slices from transgenic mice expressing the green fluorescent protein (GFP) to visualize single-fluorescent neurons and astrocytes responding to SD in real time, at high magnification and deep in intact tissue (Fig. 3) (50). We have made cerebral slices from transgenic mice expressing GFP in a small subset of pyramidal neurons in cerebral cortex and hippocampus (51) and from mice expressing GFP in a small percentage of astrocytes (52). The small number of cells producing GFP results in Golgi-like staining of neuronal somata, dendrites, and astroglia with fine astrocytic processes clearly visible. A submersion-type imaging/recording chamber (RC-29, 629 μL working volume, Warner Instruments, Hamden, CT) is mounted on the microscope stage (Luigs & Neumann, Ratingen, Germany). The aCSF is bubbled with 95% O2/5% CO2 and preheated before delivery into the chamber to prevent degassing. The slice is held down by an anchor (SHD-27LP/2, Warner) and perfused with oxygenated aCSF at 32–34°C using a recirculating system. The solution flow in and out of the chamber is controlled by two peristaltic pumps (Watson-Marlow, Wilmington, MA) set at the rate of 8 ml/min. At this rate, it takes ∼17 s to exchange solutions in the chamber. Temperature is monitored by a thermistor probe within 1 mm of the slice and maintained by an in-line solution heater/cooler (CL-100, Warner) with a bipolar temperature controller (TA-29; Warner). Evoked field EPSPs and the negative shift of extracellular potential, which signifies SD in slices, are recorded with an extracellular glass electrode (2–3 MΩ) filled with aCSF. Signals from a MultiClamp 200B amplifier (Molecular Devices, Sunnyvale, CA) are filtered at 2 kHz, digitized at 10 kHz, and analyzed with pClamp 9 (Molecular Devices).

Fig. 3.
No recovery from dendritic beading with spine loss and neuronal somata swelling after 1 h of washing out in normal aCSF following SD triggered by 100 μM ouabain application. Images are MIPs of several optical sections acquired <100 μm below the slice surface.
For 2PLSM, we use commercially available the Zeiss LSM 510 NLO META system mounted on the motorized upright Axioscope 2 FS microscope (Carl Zeiss, Jena, Germany). 2PLSM images are collected with IR-optimized 40×/0.8 NA or 63×/0.9 NA water immersion objectives (Zeiss). The scan module is directly coupled with the Spectra-Physics (Mountain View, CA) Ti:sapphire broadband mode-locked laser (Mai-Tai) tuned to 910 nm for two-photon excitation. To monitor structural changes with GFP, 3D time-lapse images are taken at 0.5–1-μm increments using 4× optical zoom, resulting in a nominal spatial resolution of 28 pixels/μm with a 63×/0.9 NA objective (12 bits/pixel, 2.24 μs pixel time). As configured with a 63×/0.9 NA objective by utilizing the full width at half maximum of a point spread function measured with subresolution beads, the microscope has the resolution of 0.38 μm in the lateral dimension and 1.950 μm in the axial dimension. Emitted fluorescence is detected by internal photomultiplier tubes (PMTs) of the scan module with the pinhole entirely opened or in a whole-field detection mode by external non-descanned detectors (Zeiss). Data acquisition is controlled by Zeiss LSM 510 software.
We use the LSM 510 Image Examiner software (Zeiss) together with NIH ImageJ (http://rsb.info.nih.gov/ij/) and Bitplane Imaris software (St. Paul, MN) for volume rendering and image analysis. Some images are processed with the Scientific Volume Imaging (Hilversum, the Netherlands) Huygens Professional image deconvolution software before obtaining dendritic spine measurements. We apply a median filter (radius = 2) to reduce photon and PMT noise in images selected for presentations.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


