CHAPTER 6 Recognizing and Treating Pain in Horses
Attitudes, opinions, and therapeutic approaches concerning the treatment of pain in horses continue to evolve.1 Traditionally, pain was used as a diagnostic and prognostic tool that helped determine the source and severity of disease (e.g., articular damage, laminitis, abscess, colic), More recently, however, pain has become appreciated for its ability to produce profound behavioral, neuroendocrine, metabolic, and immunologic effects. Chronic pain, for example, can produce avoidance and protective behaviors, poor performance, inappetence, and weight loss and predisposes to infection, resulting in a generalized “sickness syndrome,” which often contributes to the death or euthanasia of the horse.2 Improved knowledge of the neuroanatomic and molecular mechanisms responsible for pain and the discovery that untreated pain induces changes in the central nervous system (CNS) that contribute to the development of chronic pain have focused attention on the development of more aggressive and effective analgesic protocols for horses.3–5 Pain in horses should be considered an independent, rather than dependent, variable particularly when the cause of pain is uncertain (idiopathic pain), the pain is difficult to treat (e.g., myopathy or uveitis), or the cause of pain is impossible to eliminate (e.g., laminitis or osteoarthritis). Pain, like heart rate, respiratory rate, and temperature, should be considered a clinical sign that requires evaluation, documentation, and treatment. This approach will hasten the development of practical and clinically relevant diagnostic and evaluative methods that will aid in identifying the most efficacious therapies. This chapter presents an abbreviated review of the physiologic and pathophysiologic mechanisms responsible for pain and suggests clinically useful methods for evaluating and treating pain in horses.
PAIN
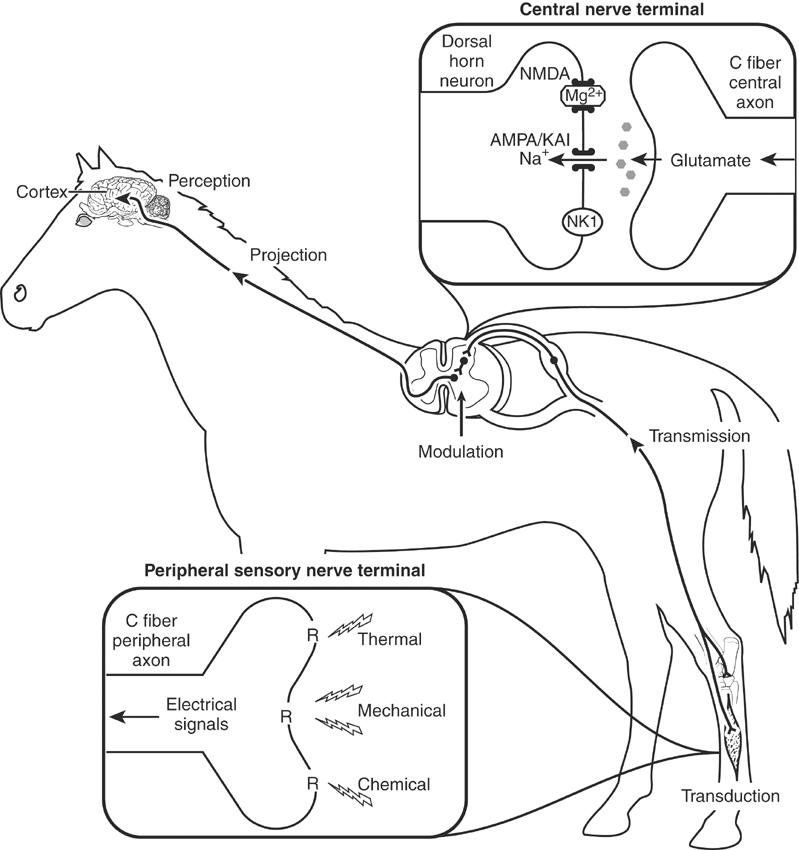
Pain is defined as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage”6 (Box 6-1). This definition encompasses those neurophysiologic processes that warn and protect the horse from actual or potential tissue damage, help prevent further injury, and promote healing. Clinically, pain results from two multifaceted components, nociception and perception.4 Both facets are integrated to provide the immediate recognition and elimination (if possible) of the painful stimulus. Nociception is the detection of a noxious (mechanical, thermal, chemical) stimulus and by itself is not pain, because the transformation of noxious stimuli to electric impulses (transduction), their transfer to the spinal cord (transmission), and their eventual projection to the brain do not ensure that the electric impulses will be recognized (perceived; e.g., as during anesthesia; Figure 6-1). Perception is the recognition that nociception has occurred or continues to occur and triggers responses that protect the horse from further insult and help maintain homeostasis. Pain perception or the actual registration of a noxious stimulus depends on external (environmental) and internal (patient-related) issues and is responsible for the secondary behavioral (vigilance, immobilization, fear), metabolic (hyperglycemia), neuroendocrine (cortisol, catecholamines), and physiologic (heart rate, respiratory rate, pupillary) responses that are frequently used to evaluate the severity of pain, determine stress, and ultimately ascertain the horse’s quality of life.
BOX 6-1 TERMS USED TO DEFINE AND DESCRIBE PAIN
Physiologic and Pathologic Pain
Under normal circumstances pain is caused by the activation of high-threshold (high stimulus intensity) pain receptors (nociceptors) located at the distal ends of unmyelinated (C) or poorly myelinated (Aδ) nerve fibers2,4 (see Figure 6-1). The free nerve endings of these peripheral afferent nerve fibers encode noxious stimuli depending on the modality, intensity, duration, and location of the stimulus. The intensity of the stimulus required to produce painful sensations is considerably more than that required to produce nonpainful sensations (touch) and is the most important factor determining the severity of pain.2 Pain that is not associated with tissue damage is referred to as physiologic or nociceptive pain and serves to warn and protect the horse from potential tissue damage. Pain that occurs in association with tissue or nerve damage is considered pathologic and is frequently categorized as inflammatory or neuropathic, depending on the primary causative factor.5 Pathologic pain is exaggerated by the development of peripheral and central sensitization. The temporal aspects of pathologic pain are dynamic and characterized by a reduction in the intensity of the stimulus required to initiate pain (hypersensitivity), the persistence of pain after removal of the noxious stimulus (hyperpathia), and the sensation of pain by stimuli that do not normally provoke pain (allodynia).7 Inflammatory and neuropathic pain are often characterized by an exaggerated response (hyperalgesia) when a noxious stimulus is applied to the injured area.
Peripheral Sensitization
Peripheral sensitization is caused by the production and activation of enzymatic and reparative processes initiated by tissue damage and inflammation at the site of injury. The production or dissemination of the by-products of damaged tissues, the infiltration and activation of inflammatory cells (lymphocytes, neutrophils, mast cells, macrophages), and increased sympathetic nerve activity excites and increases the sensitivity (lower activation threshold) of peripheral nociceptors (Figure 6-2).4,8 Tissue damage results in the production of prostaglandins, bradykinin, and neurotrophic growth factors, the local release and spread of adenosine, adenosine triphosphate and ions (H+, K+), and an exaggerated pain response. Leukocytes and macrophages release cytokines (interleukin-1 [IL-1], IL-6, tumor necrosis factor). Primary afferent sensory nerve fibers release neuropeptides (substance P, calcitonin gene-related peptide) causing mast cells to degranulate, local vasodilation, and plasma extravasation, further amplifying the inflammatory response. Collectively, this “sensitizing soup” lowers nociceptor threshold and activates additional, so-called silent, nociceptors, thereby amplifying the pain response.8 Peripheral sensitization is responsible for primary hyperalgesia or pain that is initiated by innocuous stimuli applied within the area of tissue injury.
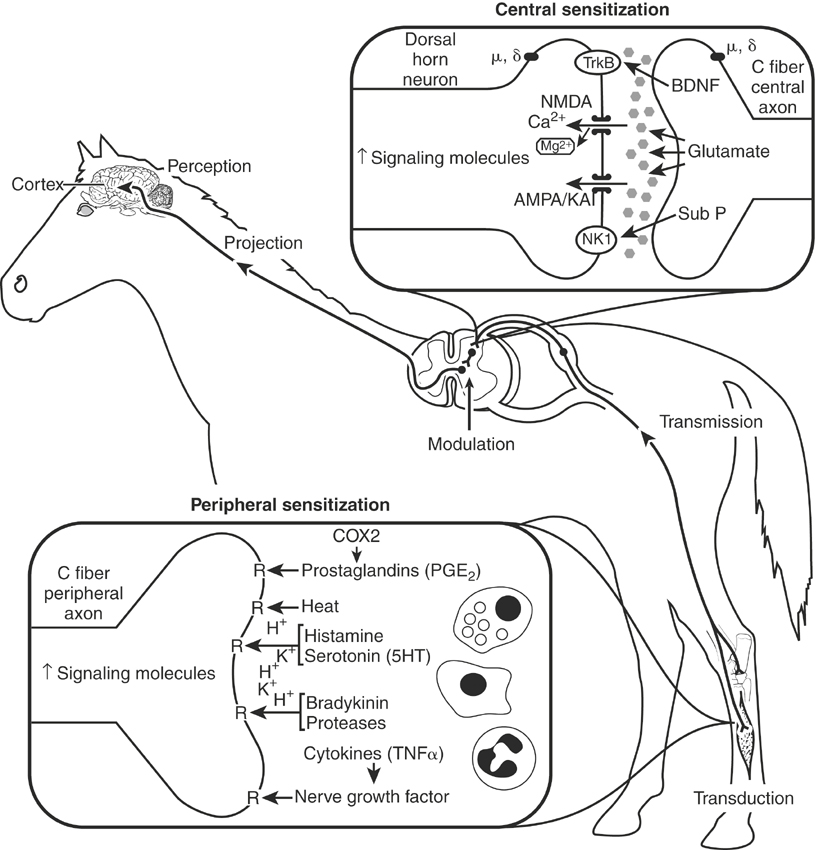
Central Sensitization
Central sensitization results from the cumulative effects (temporal summation) of repetitive and sustained nociceptive inputs on the dorsal horn of the spinal cord.4,7,8 Normally, temporary noxious stimuli generate fast but short-lived excitatory potentials within the CNS that indicate the onset, duration, intensity, and location of the painful stimulus. This fast excitatory transmission is transmitted by Aδ and C fibers and is mediated by glutamate acting on α-amino-3-hydroxy-5-isoxazole proprionic acid (AMPA) and kainite (KA) receptors within the dorsal horn of the spinal cord. Sustained input from damaged tissue or injured nerves, however, releases neuropeptides (substance P, neurokinin A) in addition to glutamate that activate N-methyl-d-aspartate (NMDA) and tachykinin receptors, resulting in a gradual “windup” of neurons in the dorsal horn of the spinal cord and central sensitization (see Figure 6-2). 8 Windup, central sensitization, and biochemical changes that occur in the CNS associated with their development represent a continuum of the pain process that is initiated by an acute painful process or untreated acute or chronic pain. Central sensitization can last for hours to days and is believed to be responsible for increases in the receptive field and hypersensitivity to noxious stimuli outside the area of primary tissue damage (secondary hyperalgesia).8,9 Changes in spinal cord segmental inhibitory input also contribute to dorsal horn hyperexcitability, resulting in the perception of pain from otherwise innocuous stimuli carried by Aδ (touch) nerve fibers.8 Central sensitization is fundamentally different from peripheral sensitization in that it enables low-intensity stimuli to produce pain because of changes in sensory processing in the spinal cord. The projection of electric impulses, initiated by a painful stimulus from the spinal cord to the brain, can modify memory and may be the reason that severe or untreated acute or chronic pain results in permanent behavioral changes.10 The development of central sensitization has several clinically relevant implications, including a change in the therapeutic focus from peripheral (local anesthetics, nonsteroidal antiinflammatory drugs [NSAIDs]) to central sites (opioids, α2-adrenoceptor agonists), the selection of potent centrally acting analgesic drugs, and the introduction of strategies that help limit or prevent the development of central sensitization (preemptive and multimodal).5 Once hyperexcitability (central sensitization) is established, larger doses of analgesics are required for longer periods of time for therapy to be effective. In other words, pain should be treated as early as possible and preemptively when possible.
In summary, peripheral sensitization is caused by the local production, release, and accumulation of chemicals (prostaglandins, leukotrienes, neuropeptides, nerve growth factors) that sensitize peripheral nerve fibers and activate additional (“silent”) pain receptors.4,8 Some of the hypersensitivity at the site of injury and all of the hypersensitivity outside the site of injury (secondary hyperalgsia) result from central sensitization.7–11 The primary difference between peripheral and central sensitization is that the former activates and sensitizes nociceptors at the terminal ends of Aδ and C fibers, whereas the latter changes sensory processing in the spinal cord, resulting in pain from stimuli that activate low-threshold sensory (touch) receptors.5,8 Visceral pain (e.g., pain from intestine, liver, spleen, kidney, or bladder), unlike somatic pain, is transmitted by parasympathetic (principally vagal) and sympathetic splanchnic afferent nerve fibers.12,13 The autonomic fibers involved in transmitting noxious inputs from visceral organs, including the distal colon, rectum, and bladder, are diffuse and extensively overlap. These differences from somatic transmission (transmission by parasympathetic and sympathetic nerves and extensive overlap) result in a significant autonomic response (tachycardia, tachypnea, mydriasis) and difficulty in localization of the site of pain.12 Generalized or diffuse inflammation, ischemia, and mesenteric stretching or intestinal dilation (e.g., of the stomach or cecum) can produce severe, unrelenting pain in horses.
STRESS AND DISTRESS
Pain associated with disease, accidental, or surgical trauma or originating from unknown causes can result in activation of the sympathetic nervous system, secretion of glucocorticoids (primarily cortisol), hypermetabolism, sodium and water retention, and altered carbohydrate and protein metabolism. The term given to this response is stress, which encompasses the biologic processes whereby the horse attempts to maintain homeostasis and cope with threats to its well-being.14 Acute and chronic pain are capable of producing a significant stress response in horses, thereby reducing their quality of life. The stress response is an adaptive pattern of behavioral, neural, endocrine, immunologic, hematologic, and metabolic changes directed toward the restoration of homeostasis (Figure 6-3). During threatening circumstances the stress response prepares the horse for an emergency reaction that fosters survival in the face of immediate threats (fight or flight). Most stress in horses is temporary because of the removal or short duration of exposure to the stressor. Surgery and anesthesia are common sources of stress in horses that can be modified by appropriate selection and use of preanesthetic and anesthetic drugs.15 Suffering occurs when horses are forced to endure the infliction or imposition of physical pain or develop the feeling of impending harm (fear).16 Severe or chronic stress can become maladaptive, producing distress and triggering self-sustaining cascades of neural and endocrine responses that upset physiologic homeostasis and negatively affect biologic functions critical to the well-being of the animal (Box 6-2). Stress and distress negatively affect the well-being of horses, produce immune incompetence, and can lead to gradual physical deterioration and death.17 At a minimum, all animals should be granted the “five freedoms”: freedom from hunger and malnutrition, freedom from discomfort, freedom from disease, freedom from injury, and freedom from pain.
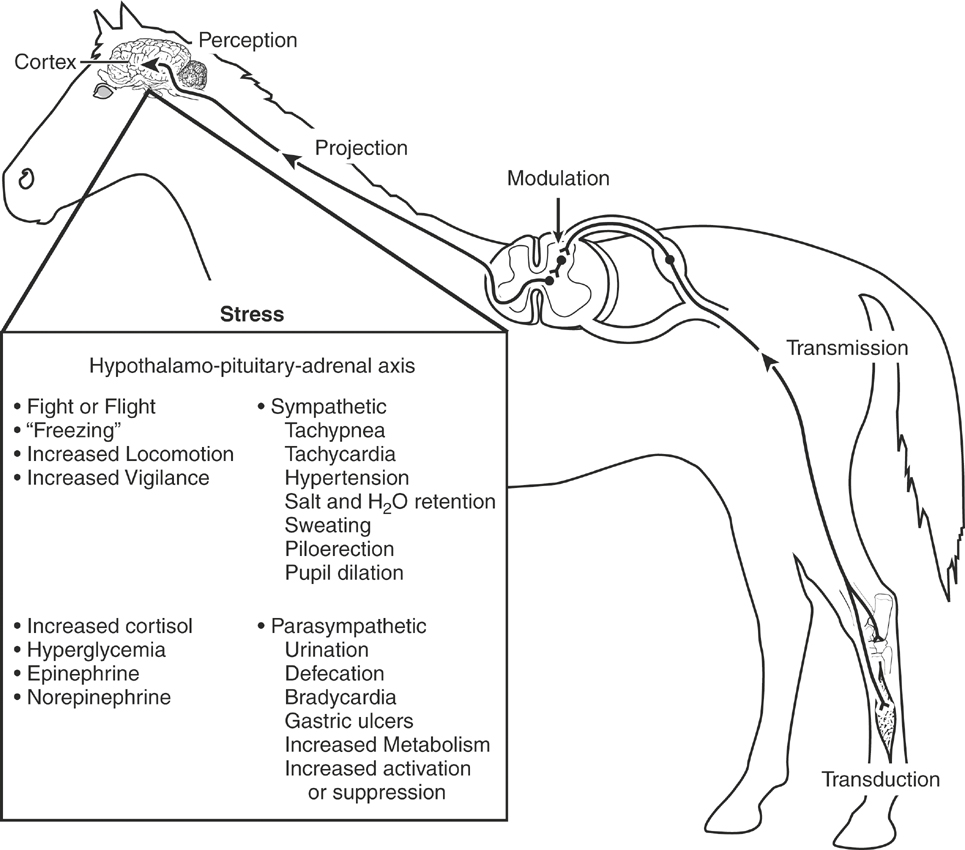
Neuroendocrine and Autonomic Stress
The noxious stimuli that produce acute and chronic pain induce stress that activates the hypothalamus-pituitary-adrenal axis.14 This afferent information stimulates the secretion of corticotropin-releasing factor, vasoactive intestinal peptide, and adrenocorticotropic hormone (ACTH). Corticotropin-releasing factor acts synergistically with vasopressin to stimulate the production of ACTH and β-endorphin, thereby enhancing cell survival and producing analgesic effects, respectively. Corticotropin-releasing factor also stimulates the adrenomedullary release of ACTH and catecholamines. Corticotropin-releasing factor is an excitatory neurotransmitter in the locus ceruleus that increases norepinephrine release and excitatory behaviors. Adrenocorticotropic hormone stimulates the adrenal cortex to secrete cortisol, corticosterone, and aldosterone and also stimulates increased glucocorticoid production and the adrenomedullary secretion of catecholamines. Cortisol stimulates gluconeogenesis, increases proteolysis and lipolysis, facilitates catecholamine effects, and produces antiinflammatory actions. It is important to note that many of these same factors (corticotropin-releasing factor, ACTH, and corticosterone) are significant modulators of learning and memory processes.18 Increases in CNS sympathetic output increase respiratory rate, heart rate, and arterial blood pressure and produce sweating, piloerection, and pupillary dilation. Catecholamines are released from the adrenal medulla into the systemic circulation, amplifying these effects. Increased concentrations of circulating catecholamines augment glycogenolysis, increase gluconeogenesis, inhibit insulin release, and promote insulin resistance. Skeletal muscle blood flow generally increases out of proportion to increases in blood flow to other organ systems, preparing the animal for fight or flight. Epinephrine causes glycogenolysis and gluconeogenesis and inhibits insulin release while promoting peripheral insulin resistance and lipolysis. Thyroid hormones stimulate carbohydrate metabolism and heat production and increase and sensitize α-adrenergic receptors in the heart, thereby sensitizing it to the effects of circulating catecholamines.



