Fig. 8.1.
Genetic architecture of common psychiatric disorders. Based on current findings, genetic variants that are relatively common in the population (>5%) have minimal impact on disease risk with most of these variants having low odds ratios (OR). In contrast, the rare genetic variants (<1%) so far identified have a more substantial impact on disease risk with most having high OR.
2.1 The Impact of Common Genetic Variants
Modern association studies exploit ancestral genetic variants that are now common (>5% minor allele frequency) in the population. These studies can focus on candidate genes based on a priori evidence or identify candidates in an unbiased manner using genome-wide association (GWA) studies (4). So far, based on the CDCA hypothesis, more than 1400 studies have investigated nearly 800 candidate genes for association with schizophrenia (http://www.szgene.org). Many of these are considered strong susceptibility genes, but none have unequivocal statistical support (5, 6). Combined, well-powered, GWA studies of psychiatric disorders have identified two candidate loci for bipolar disorder (10q21.2 and 12p13.3) (7), one candidate locus shared between bipolar disorder and schizophrenia (10q26.13) (8), and one locus for autism (15p14.1) (9). It is interesting to note that these loci do not include any top candidate genes.
While there is some debate about the relative merits of candidate gene versus GWA approaches, both suffer from similar limitations. They both use a restricted set of polymorphic markers that may or may not be linked with other unseen variants. Given that the markers genotyped represent a small fraction of all common genetic variants, significantly associated markers in GWA studies are a priori unlikely to be the true causative variants (10). Moreover, these variants often occur in regulatory regions whose effects are currently unknown or lie distal to genic regions leaving open the question of which genes are affected. Thus, even if statistically unequivocal common risk alleles are found in the near future, these are likely far removed from the actual causative allele. Follow-up studies, including replication and fine mapping studies, may help in identifying the causal alleles, but even then the functional impact of these may remain uncertain (11).
2.2 The Impact of Rare Genetic Variants
The rarity of families afflicted with psychiatric disorders in a strictly Mendelian fashion limited, until recently, acceptance that rare mutations play a significant etiological role. Taking into account variable penetrance and de novo occurrences, however, rare alleles may contribute substantially to both familial and sporadic cases (12–14). It is not surprising that the alleles with strongest statistical support for association with schizophrenia are rare, highly penetrant structural mutations. These include copy number mutations of 22q11.2 (14–16) and possibly 1q21.1, 5q13.3 (14, 16) as well as a balanced chromosomal translocation t(1;11)(q42.1;q14.3) (17). Moreover, recent surveys indicate that rare copy number mutations as a whole are over-represented in cases of schizophrenia and autism (13, 14, 18). These data suggest that rare alleles contribute substantially to the risk of psychiatric disorders.
Unlike common variants, a rare allele may be population, family, or even individual specific. In the latter case, it is not possible to prove unequivocally that the mutation is pathogenic since individuals carry many private mutations. It is possible, however, to determine whether cumulative mutational load for a given locus or biological pathway is greater for affected versus unaffected individuals collectively (4). While current estimates indicate that only a minority of cases (≤1%) are attributable to any given rare, recurrent allele (14, 16), this is likely to be a lower limit. Improvements in the resolution and efficiency of genotyping platforms will increase detection of rare alleles and their association with disease. Moreover, common variants account for only a small proportion of familial risk for common diseases, leaving the possibility that rare alleles contribute significantly to the remaining risk (19). Nevertheless, it is possible that the penetrance or the expressivity of rare mutations is determined by common genetic variation (11).
2.3 Implications for Mutant Animal Models
In order for mutant animal models to uncover a genuine pathogenetic link between a candidate gene and disease risk, the association of the genetic variant must be unequivocal, its functional effect known, and the risk allele modeled accurately. None of these conditions are met for current candidate genes or variants identified through GWA studies and thus the prospect of creating valid animal models based on common alleles is strikingly low (2). Unlike association studies, assessments of rare variants identify specific, and very likely causative, disease-associated alleles. These rare alleles are far more likely to have deleterious functional consequences than are common alleles due to clear effects on gene products. As a result they have higher potential for revealing insights into disease etiology than do less penetrant common alleles (12). In fact, the few candidate genes with common alleles of clear functional effect (e.g., COMT rs4680 and BDNF rs6265 ) show the most inconsistent clinical associations (20, 21). Therefore, because the evidence of disease association for rare alleles is often more statistically sound and the effects of the mutation more transparent, it is possible to engineer genetically faithful, and thus etiologically valid, animal models.
3 Animal Models of Disease-Associated Loci
Although the selection of candidate genes is controversial and their validation problematic, it is possible to dissect the functional roles of candidate genes and investigate the biological pathways in which they participate. This allows the generation of hypotheses regarding how these pathways may dysfunction and give rise to psychiatric disorders. When interpreting results from animal models of candidate genes, it is important to realize that the selection and the verification of candidate genes are not always independent of each other, making gene validation a potentially circular and self-bootstrapping process. Moreover, the fact that behavioral tests used to characterize models lack specificity raises the issue of whether a suite of behavioral changes can validate a mutant model in the absence of strong corroboration from human genetics.
In this section we describe mutant animal models from four candidate genes, including three genes found in genomic loci showing genetic linkage with schizophrenia (NRG1, DTNBP1, and PPP3C). While linkage signals may reflect rare as well as common alleles, current technology allows only the efficient genotyping of common makers. Thus, association studies that follow-up linkage results assume a significant contribution of common variants within these loci.
3.1 Neuregulin 1 (NRG1) Susceptibility Locus
A significant linkage peak at 8p22 implicated Neuregulin 1 (NRG1) as a schizophrenia susceptibility locus. Despite a lack of robust statistical support for specific alleles, NRG1 and its receptor, ERBB4, are among the leading candidate susceptibility genes for schizophrenia (22, 23). NRG1 is a trophic factor that, through binding of receptor tyrosine kinases, is involved in cellular proliferation, migration, neurite outgrowth, synaptic transmission, and intracellular signaling (24). This high level of pleiotropy suggests that genetic variants affecting NRG1 signaling could influence disease risk through any number of biological pathways.
Several groups have created mutant animal models affecting various domains of the Nrg1 gene to assess their impact on behavior. The first group to describe an Nrg1 mutant mouse deleted the EGF-like domain of the encoded protein and showed that these mice displayed increased locomotor activity in multiple contexts but intact short-term memory (25). Stefansson et al. (22) analyzed mutant mice lacking the transmembrane domain of Nrg1 and found that they had increased locomotor activity in an open field and impaired sensorimotor gating as measured with PPI, but normal anxiety-related behaviors. These mice were further analyzed to show abnormal social interactions but normal short-term spatial memory (26, 27). Rimer et al. (28) targeted a subset of Nrg1 isoforms by deleting the Ig domain and found that these mice showed impaired latent inhibition, but normal locomotor activity and short-term memory.
Other mutant animal models have focused on Nrg1 receptors. Similar to Nrg1 hypomorphs, homozygous ErbB4 knockout mice, but not ErbB2 or ErbB3 knockouts, showed increased locomotor activity and impaired PPI. Roy et al. (29) used a transgenic approach to express a dominant-negative ErbB4 receptor specifically in oligodendrocytes. In addition to decreased complexity of oligodendrocytes and reduced myelin thickness, these mice showed decreased locomotor activity, increased anxiety-related behaviors, abnormal social interactions, and hyperactive midbrain dopamine function. Barros et al. (30) crossed floxed ErbB2 and ErbB4 mice with CRE-expressing mice to knock out ErbB2/4-mediated Nrg1 signaling within the CNS and showed that these mice had increased locomotor activity, increased aggressivity, and decreased PPI. Surprisingly, cortical development of these mice was normal. They did, however, show decreased spine maturation of both neocortical and hippocampal neurons.
Savonenko et al. (31) investigated Nrg1 function by assessing knockout mice of one of its proteolytic processors, Bace1, which were previously used as an animal model of Alzheimer’s disease prevention (32). These mice showed increased locomotor activity in several contexts, impaired PPI, abnormal social interactions, impaired fear-associative memory, and impaired spatial short-term memory, but normal long-term spatial reference memory. Mutant mice also showed changes in dendritic complexity and spine density of hippocampal neurons. It is unclear, however, if these effects were due to Nrg1 or one of the other downstream substrates of Bace1.
Overall, these studies show that disruptions in Nrg1 signaling affect locomotor activity and sensorimotor gating but have less reliable impact on cognitive function. Given that there is, as of yet, no clear disease-associated functional allele within the NRG1 locus, it is uncertain which, if any, of the current mutant animal models are relevant to schizophrenia.
3.2 Dystrobrevin-Binding Protein 1 (DTNBP1) Susceptibility Locus
DTNBP1 was first identified as a candidate gene by fine mapping of a significant linkage signal at 6p22. Several subsequent association studies indicated that common variants within this locus may influence disease risk. As in the case of NRG1, however, the statistical support is equivocal (5, 6). DTNBP1 is widely expressed across tissues and organs, including the CNS, and is involved in the trafficking of lysosome-related organelles. Although disputed as a strong susceptibility locus, DTNBP1 remains a popular candidate gene and the availability of a naturally occurring null allele in a mouse strain makes it an attractive candidate to model.
The sdy strain of mice carries a spontaneous Dtnbp1 null allele resulting from a ∼38-kb deletion that abolishes production of the dysbindin-1 protein (33). These mice show systemic abnormalities in vesicle trafficking to lysosomes, resulting in albinism and pulmonary fibrosis. Takao et al. (34) carried out a behavioral screen of homozygous sdy mice and found that mutant mice had decreased locomotor activity, impaired long-term spatial reference memory, and impaired acquisition of a short-term memory task, but normal anxiety-related behaviors and intact neurological functions. Bhardwaj et al. (35) also characterized sdy mice and showed that homozygous mutant mice had decreased locomotor habituation and increased fear-associative memory. While both heterozygous and homozygous mutants showed impaired short-term recognition memory and a blunted locomotor response to acute amphetamine, only homozygous mice were hypersensitized to amphetamine after chronic treatment. Feng et al. (36) examined homozygous sdy mice and found they had normal locomotor activity and normal anxiety-related behaviors but impaired social interactions and long-term recognition memory. Cox et al. (37) backcrossed sdy mice into a C57BL/6 background and used wild-type littermate controls. Homozygous mutant mice showed increased locomotor activity, decreased locomotor habituation, impaired spatial navigation, and impaired long-term spatial reference memory but less anxiety-related behaviors and improved motor learning. Given its role in vesicle trafficking, it is not surprising that dysbindin mutant mice show deficits in neurotransmitter release (38). These deficits as well as dopamine dysfunction in these animals (39) may be relevant to the putative role of DTBNP1 in schizophrenia.
Although methodological differences, such as the use of littermate controls, may account for contradictory findings from sdy mice, further studies are needed to confirm dysbindin’s role in the CNS and define its neurobiological functions. Like NRG1, however, the lack of clear disease-associated, functional alleles within the DTBNP1 locus makes dysbindin’s role in disease risk unclear. Thus, even though there is evidence of reduced dysbindin expression in schizophrenic brain tissue (40), this makes the relevance of sdy mice to schizophrenia pathogenesis uncertain.
3.3 Other Susceptibility Loci
Near the 8p22 linkage peak that implicated NRG1 as a candidate gene resides PPP3CC, which encodes for a calcineurin catalytic subunit. There is some support that common variants within this locus are associated with schizophrenia (41). Mice with a conditional knockout of calcineurin’s regulatory subunit provide additional support because they display behavioral deficits similar to animal models of other schizophrenia candidate genes. These deficits include increased locomotor activity, decreased PPI, impaired short-term spatial memory, and impaired reversal learning (42, 43). Variants within the regulatory subunit gene itself, however, have not been associated with schizophrenia. Nevertheless, taken together, genetic and animal model findings point to a potentially important role of this pathway in disease susceptibility, possibly via integrating glutamatergic and dopaminergic signaling as well as modulating synaptic vesicle recycling (44, 45).
AKT1, a functional candidate gene, provides another relevant example. While AKT1 was not identified from a linkage peak, AKT1 protein levels and phosphorylation of its substrate GSK3β were reduced in brains and lymphocytes of individuals with schizophrenia (46). AKT1 it is an attractive candidate primarily because of its functional role in dopaminergic signaling (47, 48). Similar to other candidate genes, the support for common variants within this locus increasing disease risk is mixed (http://www.szgene.org). Nevertheless, mice deficient in Akt1 show a greater sensitivity to the disrupting effects of amphetamine on PPI (46) and have altered dendritic complexity in medial prefrontal cortex (mPFC) and impaired working memory performance under various pharmacological challenges. Whether any other changes produced by Akt1 deficiency (48) are also relevant to disease risk remains to be determined.
4 Animal Models of Disease-Associated Alleles
The increasing ability to detect rare genetic lesions strongly associated with psychiatric disorders provides a unique opportunity for generating etiologically valid mutant animal models. In this section we discuss animal models of the strongest inherited and de novo genetic risk factors for psychiatric disorders known: a familial truncation of the DISC1 gene and sporadic copy number mutations of 22q11.2, respectively. Although these lesions are individually rare, the ability to model them accurately allows identification of the exact cellular pathways and neural circuits likely involved in disease pathogenesis.
4.1 The Disrupted-in-Schizophrenia 1 (DISC1) Susceptibility Locus
DISC1 was identified through a balance chromosomal translocation t(1;11)(q42.1;q14.3) segregating with schizophrenia and mood disorders in a large Scottish pedigree. Like NRG1, DISC1 is a highly pleiotropic gene with effects on cellular proliferation, migration, dendritogenesis, synaptogenesis, and intracellular signaling (49). Association studies suggest that common variants within the DISC1 locus may influence the risk of psychotic and mood disorders in general (50). Like other association studies, however, these studies of DISC1 have not produced any statistically unequivocal risk alleles. This means the well-defined mutation in the Scottish family is currently the only disease-associated allele within the DISC1 locus capable of being modeled validly in animals.
While the biology of DISC1 is being interrogated with many analytical tools, such as shRNA (51–53), these are not meant to recapitulate the integrated effects of a genetic lesion. Thus, the relevance of these findings to schizophrenia pathogenesis remains uncertain and, in fact, has produced opposing results from animal models carrying germline mutations (54). We therefore mainly discuss DISC1 mutant models that have attempted to recapitulate the Scottish mutation. We summarize the main findings from these models with a focus on the different methodological approaches used by investigators.
Koike et al. (55) were the first to describe a DISC1 animal model. They used a knock-in approach to introduce a truncating lesion in the endogenous murine Disc1 orthologue to resemble the putative effects of the translocation (Fig. 8.2, Disc1 tm1Kara ). This abolishes expression of the major Disc1 isoforms and, despite an artificial polyadenylation signal, causes low expression of the truncated protein, indicating its instability under physiological conditions (54). These mutant mice showed no changes in locomotor activity or PPI (55). They did, however, show a unique profile of cognitive impairments. A battery of tests revealed no changes in fear-associative, object recognition, long-term spatial reference, or short-term spatial memories (54). Rather, both heterozygous and homozygous mutant mice exhibited consistent impairments in working memory tests (i.e., short-term memory tests with high demands on cognitive control) (54, 55). Interestingly, the cognitive deficits in mutant mice were in the context of minimal histopathological changes in mPFC, with more evident changes in the hippocampus including changes in the distribution and organization of young and mature dentate gyrus granule cells. While the cognitive profile of translocation carriers is unknown, the cognitive impairments in these mice may relate to the cognitive control deficits prominent in psychotic and mood disorders.
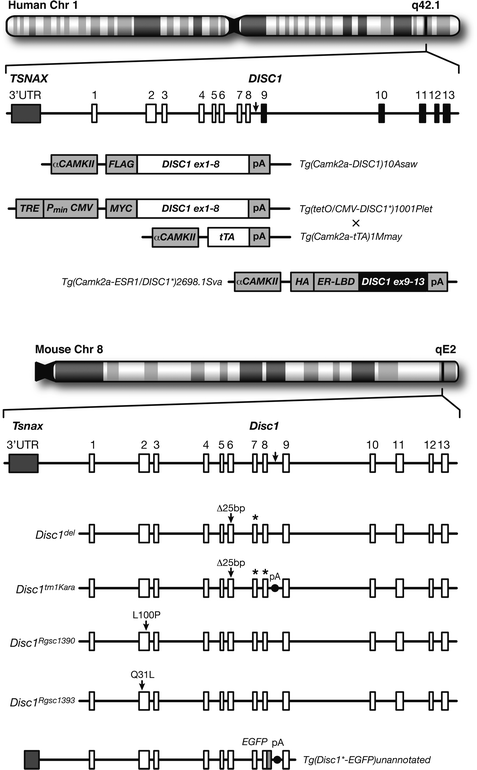
Fig. 8.2.
Animal models of the DISC1 locus. (Top) Chromosomal location and genetic structure of the human DISC1 locus. The t(1;11) translocation break point occurs between exons 8 and 9 (arrow) with exons 9–13 (black) relocated to chromosome 11. Below it are the allele symbols and transgenic constructs used to model the functional effects of the translocation. These models interfere with endogenous mouse Disc1 function in a dominant-negative [Tg(Camk2a-DISC1)10Asaw and Tg(tetO/CMV-DISC1 * 1001Plet + Tg(CAmk2a-tTA)1Mmay] or transdominant-negative [Tg(camk2a-ESR1/DISC1 * )2698.1Sva] manner. (Bottom) Syntenic chromosomal location and genetic structure of the mouse Disc1 locus and the corresponding break point location (arrow). Below it are the various mouse Disc1 alleles so far described. Disc1 del arose from a spontaneous 25-bp deletion within exon 6 that introduces a premature stop codon in exon seven (*) of 129 and related strains of mice. Disc1 tm1Kara was engineered to carry a premature stop codon at the end of exon 8 (*) followed by a polyadenylation signal in an attempt to recapitulate the translocation, but since this was created in a 129S6/SveV background, the allele also carries the endogenous 25-bp deletion. Disc1 Rgsc1390 and Disc1 Rgsc1393 have missense mutations in exon 2 identified through a mutagenesis screen and mice carrying these alleles show divergent behavioral phenotypes. Although not meant to model the translocation and thus not discussed in the main text, these mutations indicate that allelic heterogeneity of the DISC1 locus may account for diverse psychopathologies. Tg(Disc1 * -eGFP)unannotated is a transgenic allele constructed from an artificial bacterial chromosome consisting of exons 1–8 of the mouse Disc1 fused to enhanced green fluorescent protein (eGFP). Mice carrying two copies of this allele also have two wild-type, endogenous Disc1 copies.
Other mouse models were designed under the assumption that the (1;11) translocation produces a truncated protein and thus overexpresses a truncated form of human DISC1. Hikida et al. (56) were the first to produce such a model and used a αCAMKII promoter to drive truncated DISC1 expression in forebrain principal neurons (Fig. 8.2, Tg(Camk2a-DISC1)10Asaw). These transgenic animals showed increased lateral ventricular volume in adulthood, reduced numbers of parvalbumin-positive cells in the mPFC, abnormal locomotor activity, subtle deficits in PPI, impaired foraging, and increased immobility in the forced swim test (FST). There were no changes in measures of anxiety-related behaviors, long-term spatial reference memory, or short-term spatial memory as assessed in the Y maze. Pletnikov et al. (57) used the Tet-off double transgenic system to drive expression of truncated human DISC1 under the CMV promoter with tetracycline under the αCAMKII promoter (Fig. 8.2, Tg(tetO/CMV-DISC1 * )1001Plet). These double transgenic mice showed increased lateral ventricular volume, abnormal locomotor activity, and increased aggression in males but no changes in measures of anxiety-related behaviors, long-term spatial reference memory, olfaction, or PPI. Females did, however, show deficits in long-term spatial reference memory.
Unlike the previous studies that expressed an N-terminal fragment of DISC1, Li et al. (58) expressed a C-terminal fragment of DISC1 under the αCAMKII promoter using a single transgenic inducible and reversible system (Fig. 8.2, Tg(Camk2a-ESR1/DISC1 * )2698.1Sva). This approach was designed to transiently disrupt Disc1 interactions occurring at the C terminus. Mice briefly exposed to this truncated DISC1 peptide at postnatal day 7 showed abnormal responses to the FST, spatial working memory deficits, and decreased sociability but no changes in locomotor activity or measures of anxiety-related behaviors. Interestingly, no deficits were apparent when the mutant DISC1 fragment was expressed during adulthood.
It is important to note that the above transgenic models do not necessarily recapitulate the effect of the disease-associated allele found in the Scottish family. The ectopic expression of mutant DISC1 does not reflect Disc1 expression in terms of absolute levels or spatial and temporal patterns. In an attempt to circumvent these limitations, Shen et al. (59) engineered transgenic mice to express two copies of truncated mouse Disc1 using a BAC system, thus, presumably, preserving the temporal and spatial profiles of Disc1’s endogenous expression (Fig. 8.2, Tg(Disc1 * -EGFP)unannotated). These mice showed increased lateral ventricular volume, decreased neuronal proliferation, thinner cortices, agenesis of the corpus callosum, reduced parvalbumin-positive cells in the PFC and the hippocampus, increased immobility in the FST, and abnormal latent inhibition. While presumably the ratio of mutant to normal Disc1 copies is similar in this model to that in translocation carriers, the absolute dosage is increased by twofold and altered function of N-terminal interacting partners is possible. Although this model has advantages over other transgenic approaches, further clinical characterization of translocation carriers (e.g., MRIs) is needed to determine how accurately this model reflects the human condition.
4.2 The 22q11 Susceptibility Locus
Recent studies of human genetic variation indicate that large-scale genomic structural rearrangements are common in the population. One of the most common abnormalities is de novo microdeletion of the 22q11.2 locus. Carriers of this microdeletion are at inordinately high risk of developing schizophrenia with odds ratios exceeding ∼25-fold. Currently, 22q11.2 microdeletions represent the only confirmed recurrent mutation responsible for introducing sporadic cases of schizophrenia into the population. The most common deletion spans 3 Mb, but a nested 1.5 Mb deletion seems to be the schizophrenia critical region (15). Premorbid children with the microdeletion present with variable symptoms including impairments in attention, memory, and cognitive control, suggesting that these cognitive deficits reflect genetic liability to schizophrenia.
Fortunately, the human 22q11.2 locus is conserved within the syntenic region of mouse chromosome 16 and harbors nearly all orthologues of the human genes (Fig. 8.3). This provides a unique opportunity to create mutant animal models with a high degree of etiological validity. Kimber et al. (60) were the first to behaviorally characterize a model of the 22q11.2 microdeletion. They engineered mice deficient in seven genes spanning ∼150 kb of the 1.5-Mb deletion syntenic region (Fig. 8.3, Del(16Zpf520-Slc25a1)1Awb). Hemizygous mice showed increased PPI but normal locomotor activity, anxiety-related behaviors, fear-associative memory, and neurological function. Paylor et al. (61) characterize a 22q11.2 model spanning ∼1 Mb containing 18 orthologues of the human genes within the 1.5 Mb deletion (Fig. 8.3, Del(16Es2el-Ufd1l)217Bld). They found decreased PPI and impaired fear-associative memory but no changes in measures of anxiety-related behaviors or neurological function. Long et al. (62) used mice with a hemizygous deletion spanning ∼1.3 Mb and containing all of the orthologous genes within the 1.5 Mb deletion (Fig. 8.3, Del(Dgcr2-Hira)1Rak) and found decreased PPI and neurological dysfunction but normal fear-associative memory. Finally, Stark et al. (63) also created mice with a hemizygous deletion syntenic to the human 1.5 Mb deletion (Fig. 8.3, Del(Dgcr2-Hira)2Aam). This deletion resulted in increased locomotor activity, decreased PPI, decreased fear-associative memory, and impaired acquisition of a working memory task. These behavioral changes were in the context of decreased dendritic complexity, reduced spine density and fewer excitatory inputs to CA1 hippocampal neurons.
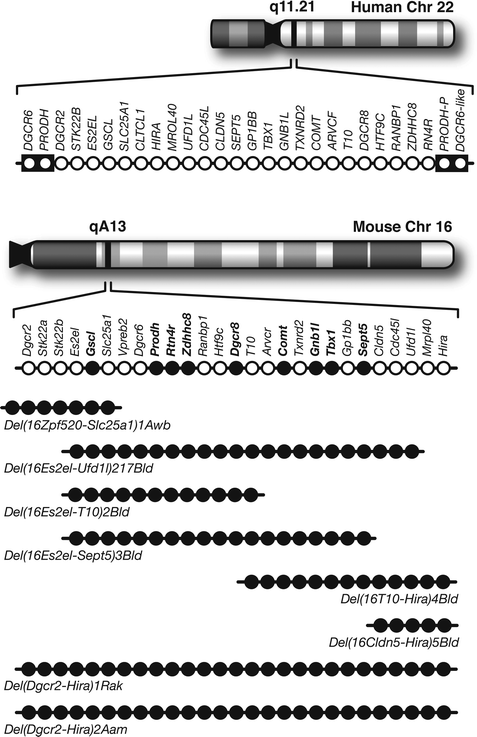




Fig. 8.3.
Animal models of the 22q11.2 locus. ( Top) Chromosomal location and genetic organization of the 22q11.2 locus. Each open circle represents one gene. This 1.5-Mb critical region is flanked by low copy repeat sequences (black boxes), making it prone to non-homologous recombination. PRODH-P and DGCR6-like are pseudogenes. (Bottom) Syntenic region of mouse chromosome 16 and the genetic organization of the corresponding orthologues. Single gene deletion models that have been analyzed with PPI are indicated in solid black circles. Below are the various multigene deletion models that have been characterized behaviorally labeled by their allele symbols. Note that only two models contain all the genes within the 1.5 Mb deletion associated with schizophrenia.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
