Fig. 14.1
The mammalian molecular clock. This circadian molecular clock has a negative feedback loop involving the genes per1-2, cry1-2, bmal1, and clock. Clock and Bmal1 are transcription factors that activate per and cry expression by binding to their E-box promoter region. Per and Cry inhibit Clock/Bmal-induced gene expression. The expression of Rev-Erba represses Bmal 1 transcription through the RAR-related orphan receptor (ROR). Picture taken from Golombek and Rosenstein (2010)
In mammals, the timing system also involves the coordination of a set of oscillators; the suprachiasmatic nucleus (SCN) serves as pacemaker, whereas the peripheral oscillators (in other brain regions as well as in thoracic and abdominal organs) play a subordinated role in basal conditions. A healthy circadian physiology depends on the correct reciprocal communication and harmonization between the SCN and the peripheral oscillators (Schibler et al. 2003).
14.3 Metabolic Regulation
The generic term “metabolism” is defined as the set of biochemical reactions that take place within the cell. It involves the synthesis and degradation of hundreds of intermediary metabolites by means of catalytic proteins known as enzymes. Metabolic networks are controlled at several levels, but three are very important: (1) energy charge, (2) redox state, and (3) cellular compartments (Liu and Zhang 1985). Energy charge refers to the proportion of adenine nucleotides within the internal milieu. The ratio ATP/AMP drives the regulation of key allosteric proteins to enhance or reduce metabolic fluxes in response to the cellular requirements to grow, differentiate, or adapt to energetic challenges (Atkinson 1968). Redox regulation concerns the regulated transit of electrons that enable the control of the oxidative–catabolic or the reductive–anabolic reactions. It is based on pairs of molecules (redox pairs) that are usually coenzymes: NAD+/NADH for degradative processes, NADP+/NADPH for synthetic pathways, and GSSG/GSH (oxidized and reduced glutathione) for modulating the redox state of protein-bound thiol groups (Veech 2006). Endomembranes make possible the formation of well-defined organelles such as mitochondria, peroxisomes, endoplasmic reticulum, and others. These intracellular compartments allow the creation of gradients of ions or metabolites that are one of the most important driving forces to control the directionality of the metabolic pathways as well as the state of membrane polarization (Elias 2010).
14.4 Overweight and Obesity
Most of the time, organisms reach equilibrium between the calories they ingest at mealtime and calories spent during their daily activities. The result is a regulated control of the body mass. However, under certain pathological circumstances this equilibrium is broken, with the concomitant consequence of under- or overweight. Obesity develops when energy intake exceeds energy expenditure. The health problem associated with obesity is not only the increase in fatty deposits in the subcutaneous white adipose tissue, but more importantly, the increases in visceral fat and the fat deposited “ectopically” in organs like the liver, pancreas, heart, or skeletal muscles that tend to cause a pro-inflammatory condition with negative health consequences (Item and Konrad 2012).
Obesity is an organic condition caused by multiple factors including genetic predisposition, altered endocrine signaling, and metabolic abnormalities; however, in order to be expressed, it requires an environmental influence, usually a change in the daily access to calories or performance of physical activity (Faith and Kral 2006).
To gain more understanding regarding the metabolic, physiological, and genetic mechanisms that underlie obesity, several experimental models and approaches have been implemented (reviewed in Kanasaki and Koya 2011). Experimental models of obesity in mice are divided into monogenic and polygenic. Among monogenic mice, the most popular are those with defects in leptin signaling, the ob/ob and db/db mice that lack the gene for leptin or its receptor, respectively. The altered aguti gene/protein (lethal yellow mutant) was the first obesity gene characterized at the molecular level more than 20 years ago. Although monogenic models provide important information about the biology of obesity, human obesity is most likely mediated by multiple genes. Therefore, polygenic models could be much more relevant to human obesity. The New Zealand obese mouse exhibits hyperphagia, reduced energy expenditure, and type 2 diabetes (only in males). The Tsumura Suzuki obese diabetic mouse shows marked hyperglycemia and hyperinsulinemia, and eventually displays lesions similar to diabetic nephropathy and neuropathy. M16 mice exhibit hyperphagia, hyperinsulinemia, and hyperleptinemia. Kuo Kond mice also show type 2 diabetes with a marked insulin resistance preceding the onset of obesity. Neotomodon alstoni is a Mexican mouse that spontaneously develops overweight and obesity when fed with a regular diet for laboratory rodents, especially the females. These mice show modified circadian rhythmicity and altered parameters related to the metabolic syndrome (Carmona-Alcocer et al. 2012).
Three types of rat have been used as experimental models for obesity: Zucker, Wistar fatty, and Otsuka Long Evans Tokushima fatty. All of them show abnormalities in glucose handling and are prone to the diabetogenic condition. Usually, the males are more likely than females to develop more severe anomalies.
Manipulating the dietary input of calories is also a very popular way to produce obese animals in biomedical laboratories. Depending on the rodent strain, a high-fat diet induces overweight and abnormalities similar to human metabolic syndrome. C57BL/6J mice show more severe irregularities than C57BL/KsJ, whereas C57BL/J6 mice, sand mice, and spiny mice exhibit a clear obese phenotype with type 2 diabetes characteristics (Collins et al. 2004). A diet supplemented with sucrose in the drinking water has also been used to produce overweight in rats and is accompanied by metabolic alterations similar to human pathologies (Larqué et al. 2011).
14.5 Cancer
Cancer is defined as an undifferentiated cell population with abnormal metabolism and uncontrolled cell division. The role of onco-proteins and tumor suppressor proteins in proliferative signaling, cell cycle regulation, and altered adhesion is well established. Chemicals, viruses, and radiation are generally accepted as agents that commonly induce mutations in genes encoding these cancer-inducing proteins, thereby giving rise to cancer. More recent evidence indicates the importance of two additional key factors imposed on proliferating cells—hypoxia and/or lack of glucose. These two additional triggers can initiate and promote the process of malignant transformation, when a low percentage of cells escape cellular senescence. Unregulated cell proliferation leads to the formation of cellular masses that extend beyond the resting vasculature, causing oxygen and nutrient deprivation. The resulting hypoxia triggers a number of critical adaptations that enable cancer cell survival. The process of apoptosis is suppressed, and glucose metabolism is altered. Aerobic glycolysis or the Warburg effect links the high rate of glucose fermentation to cancer. Together with glutamine, glycolysis of glucose provides the carbon skeletons, NADPH, and ATP to build new cancer cells that persist in hypoxia, which in turn rewires metabolic pathways for cell growth and survival (Dang 2012). Recent investigations suggest that oxygen depletion stimulates mitochondria to compensate increased reactive oxygen species (ROS). Oxygen depletion activates signaling pathways, such as hypoxia-inducible factor 1, that promote cancer cell survival and tumor growth (Hahn et al. 1999).
There are several approaches to study the biology of cancer in experimental models (reviewed in Zhao et al. 2004): (1) use of established cell lines derived from cancerous organisms; (2) immortalization and transformation using chemical, physical, and viral agents; (3) immortalization and transformation using defined combinations of genetic elements such as oncogenes; (4) immortalization by upregulation of telomere reverse transcriptase, and (5) introduction of SV40 large T antigen with an oncogenic allele of RAS.
14.6 Metabolic Interactions
There are several examples of how metabolic networks interact with transcriptional programs to regulate circadian functions that have a direct impact on the handling of cellular energy and are affected during states of obesity and cancer (Fig. 14.2).
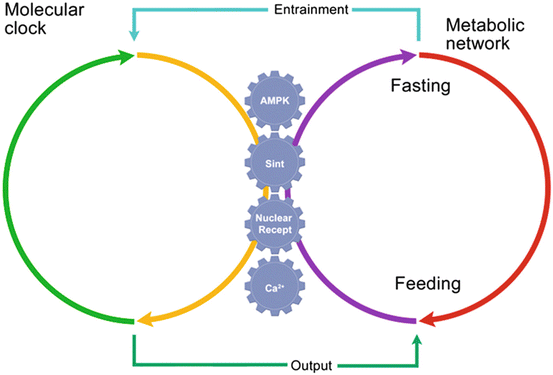
Fig. 14.2
Communication by several metabolic outputs between the molecular circadian clock and the metabolic network. The genes in the molecular clock are connected with the metabolic network by different metabolic signaling pathways; their regulation depends on whether the organism is in a fasting or feeding condition
14.6.1 Nuclear Receptors
Nuclear receptors belong to a family of transcriptional factors that recognize specific ligands, usually of lipophilic nature. These ligands can be hormones (steroid and thyroid hormones), vitamins (β-carotene and cholecalciferol and ergocalciferol), metabolic intermediates (fatty acids and derivates, bile salts, and sterols), and xenobiotics. The protein clocks ROR (α, β, and γ) and REV-ERB (α and β) are part of this family. Recent studies reveal some of the nuclear receptor genes as being direct targets of the circadian clock. Peroxisome proliferator-activated receptor-α (PPARα) and PPARγ regulate lipid metabolism and energy homeostasis by coordinated actions in a variety of tissues. Both are under the direct control of the timing system (Yang 2010). Deregulation of PPARs is involved in the onset and development of obesity (Evans et al. 2004). Nuclear receptor dysfunction has been implicated in diverse pathological states, genetic syndromes, and cancer. Co-regulators have broad physiological and pathological functions that make them promising new drug targets for diseases such as hormone-dependent cancers (Lonard and O’Malley 2012).
14.6.2 AMP-Dependent Kinase
AMPK acts as a sensor for the energy status of the cell. It is a trimeric protein that is directly regulated by the ratio AMP/ATP as well as by the cellular metabolic stress response (LKB signaling). When AMPK is active, catabolic processes are upregulated, and anabolic pathways are arrested. This enzyme induces a phase advance of circadian expression of clock genes by degrading one of the isoforms of PER (PER2) through phosphorylation of Casein kinase Iε at Ser389. AMPK also contributes to metabolic entrainment of peripheral clocks by phosphorylating and destabilizing CRY1 (Um et al. 2011). KO models of AMPK result in overweight and development of hyperinsulinemia and glucose intolerance (Steinberg et al. 2010). It has been observed that cancerous tissues show low AMPK activity, suggesting a connection among nutrient sensing, metabolic modulation, and control of cellular division in neoplasic diseases (Korsse et al. 2013).
14.6.3 Sirtuins
Sirtuins are a family of enzymes (from SIRT1 to SIRT9) that show deacetylase or ADP ribosyl-transferase activity. They are present in several compartments of the cell, including the nucleus, mitochondria, and cytosol. Their activity is dependent on the availability of NAD+, and hence, sirtuins are sensitive to the cytoplasmic and mitochondrial redox state (Denu and Gottesfeld 2012). Experimental evidence has linked the activity of sirtuins to increased longevity and the arrest of cellular decline and metabolic dysfunction associated with aging. SIRT1, through its regulation of PPAR-γ and PGC-1α activity, has a significant regulatory role in fat mobilization and fatty acid oxidation. The observation that SIRT1 can directly deacetylate core components of the circadian clock machinery is interesting, as the ultimate goal of circadian physiology is to coordinate and match intracellular metabolism to external food availability. SIRT1 is required for transcription of several core clock genes, including Bmal1, Rorγ, Per2, and Cry1 (Jung-Hynes and Ahmad 2009). Interestingly, the clock protein CLOCK has intrinsic histone acetyl-transferase activity and operates within a large nuclear complex with other chromatin remodelers. Some substrates acetylated by CLOCK are the histone H3 and the other clock protein BMAL1. SIRT1 counterbalances the enzymatic activity of CLOCK, and hence, it is an important metabolic regulator of the molecular clock (Bellet et al. 2011). Recent studies have shown SIRT1 to regulate fatty acid oxidation in the liver, sense nutrient availability in the hypothalamus, and influence obesity-induced inflammation in macrophages (Schug and Li 2011). It has been reported that SIRT3 can function as a tumor promoter (it is overexpressed in oral cancer cells and tissues) or suppressor (it is downregulated in breast cancer), depending on the cell and tumor type and the presence of different stress or cell death stimuli (Alhazzazi et al. 2011).
14.6.4 Calcium Dynamics
Intracellular calcium handling involves the coordinated action of a variety of proteins responsible for calcium mobilization, responsible for both outward and inward calcium mobilization. There are 2 principal intracellular calcium-release channels: the inositol 1,4,5-trisphosphate receptor (IP3R) (types 1–3) and the ryanodine receptor (RyR) (types 1–3). The metabolic pumps that extrude cytosolic calcium are the sarco/endoplasmic reticulum calcium ATPase (SERCA) and the plasma membrane calcium ATPase (PMCA) (Gaspers and Thomas 2005). Intracellular calcium fluctuates with different frequencies and to various extents among the cellular organelles. It has been suggested that calcium transients could be a kind of signaling code to coordinate metabolic and transcriptional activities (Berridge et al. 2000). Intracellular calcium is thought to play diverse roles as a component of the timing system (for review Báez-Ruiz and Díaz-Muñoz 2011) by regulating the entrainment process, clock gene expression, and output signaling. Diurnal fluctuations of cytoplasmic calcium as well as of RyR and IP3R have been observed in the SCN. Supporting the importance of intracellular calcium dynamics in the rhythmicity of the SCN, calcium buffered with >40 μM BAPTA-AM or with low calcium levels (Ca2+-free media) promoted damping of Per1-luc expression in SCN explants. In NIH3T3 and rat1-fibroblast cell cultures, thapsigargin and calimycin (a calcium ionophore) increased Per1 expression during the first hours of treatment. Enhanced adhesion and calcium dynamics are reported to be favorable for the invasion and extravasation required for malignant progression in prostate cancer (Bastatas et al. 2012). Munaron et al. in 2008 postulated that intracellular calcium plays a key role in tumor angiogenesis, an important step in the physiopathology of many cancerous growths.
14.7 Understanding the Mechanisms of Obesity
Obesity is currently a major health problem and is increasing worldwide; its prevalence is mainly a consequence of interactions among multiple genes, physiology, and lifestyle factors. Defined in humans as a body mass index (BMI, weight in kilograms divided by the square of the height in meters) above 30, obesity is currently associated with premature death through increased risk of many chronic diseases, including type 2 diabetes, cardiovascular disease, and cancer (Cheung and Mao 2012). For the past several years, various approaches have been used to identify the physiological and genetic bases of obesity; one of the strongest pieces of evidence involves mechanisms regulating food intake and mediated by hormones and neurotransmitters controlling the sensations of satiety in specific centers within the brain, resulting in chronic energy imbalance such that calories consumed exceed calories expended (Shin et al. 2009). This condition has also been linked to many environments that offer an abundance of calorie-rich foods and few opportunities for physical activities. Recent clinical and laboratory studies suggest that circadian biology also plays an important role in the pathogenesis of obesity, type 2 diabetes, and the metabolic syndrome (Maury et al. 2010).
Circadian rhythms are evolutionarily conserved and represent an adaptation of the organism to its environment; diurnal or nocturnal activity involves behaviors that include feeding–fasting cycles, which are also inputs to the circadian system. The circadian system in mammals is hierarchically organized the hypothalamic suprachiasmatic nucleus (SCN), identified as the dominant circadian pacemaker, imposes period and phase on peripheral oscillators, which are all cells in the body that express “clock genes.” However, internal entrainment requires a tight coordination between neural and humoral inputs, driven directly or indirectly by the SCN. These “peripheral clocks” differ in the manner in which they are reset as well as in the outputs under their control, suggesting that the system must be accurately synchronized (Mohawk et al. 2012; Menaker et al. 2013).
Studies on these peripheral clocks, especially as they affect metabolism, may give us a vision of circadian alignment for internal synchrony in normal physiology as well as in disease. The mechanisms, including this internal circadian synchrony, may include signaling at the transcriptional level, posttranscriptional, and posttranslational regulation of the proteins encoded by clock genes, and epigenetic influences of clock genes on chromatin modifications that affect gene regulation (Green et al. 2008; Bass and Takahashi 2010; Froy 2010; Bass 2012).
In normal conditions the SCN maintains temporal organization of activity, body temperature, and feeding, in such a way that local and systemic circadian signals present a particular alignment. However, when food is provided on a regular basis at a different time, out of the animal’s activity phase, anticipatory locomotor activity prior to the arrival of the meal indicates that the internal timing also depends on the coincidence of different zeitgebers, such as light and food cycles, and that a different alignment of peripheral oscillators occurs (Pezuk et al. 2012 ; Mistlberger 2011).
Unorganized activity–rest and feeding–fasting cycles in humans may cause circadian misalignment, and also may facilitate the occurrence of metabolic disorders (Scheer et al. 2009). In addition, disorders in the sleep–wake cycles are associated with irregular feeding schedules, and may be one of the multiple causes of metabolic disorders (Laposky et al. 2008).
Misalignment of the endogenous circadian phase can occur with forced or artificial non-circadian cycles that do not coincide with retinal light exposure, meal times, and the times of wakefulness/activity, work/school, exercise, and sleep; the main examples are shift work and jet lag (Bass 2012). Circadian disregulation and obesity have an initial behavioral reference; artificial bright-light exposure during the evening and hypercaloric diets consumed at night are signals that may work as “false” zeitgebers in conflict with the natural cycles. Together with reduced physical activity and sleep disorders, such behaviors favor an increase in body weight that, over a number of years, results in obesity and metabolic syndrome.
The link between obesity and clock mechanisms is just beginning to be understood. Studies performed in animal models offer unique and powerful experimental approaches and allow us to understand the underlying mechanisms of metabolic disorders and circadian disruption, as well as to develop therapeutic strategies (Arble et al. 2010). Circadian misalignment in rodents may be illustrated by experiments simulating jet lag, where some tissues are able to re-entrain to jet lag more quickly than others (Yamazaki et al. 2000). Mice with mutations in the circadian Clock gene develop obesity and metabolic syndrome (Turek et al. 2005), and genetic polymorphisms within the core clock genes Clock and Bmal1 are also associated with obesity, hypertension, and type 2 diabetes (Marcheva et al. 2010). On the other hand, just changing the feeding schedule facilitates overweight and obesity in mice. When food access occurs during the rest/fasting phase of the cycle (photophase in nocturnal rodents), a gradual increase of body weight is clearly noted in comparison with the animals that have access only during the night or ad libitum (Arble et al. 2009). In a rat model susceptible to the development of type 2 diabetes (HIP rats), disruption of circadian rhythms accelerates the loss of beta-cell function and the mass characteristic of this condition (Gale et al. 2011). Feeding time determines the targets and phases of rhythmic transcripts in liver. Thus, there is a synergistic interaction between clock and temporarily restricted feeding patterns in many important metabolic genes, including those involved in mitochondrial function. By using a Restricted Feeding protocol, it is possible to cause a completely new phase relationship between the light-entrained (SCN) and a food-entrainable oscillator (FEO). Mice and rats are naturally nocturnal feeders, and limiting food access to the light phase causes some behaviors, such as feeding-dependent hormonal rhythms and the expression of circadian clock genes and clock-controlled genes within metabolic tissues (e.g., the liver), to become entrained to the novel feeding time.
Clock gene expression in the liver is highly correlated with circadian feeding rhythms, while the SCN remains synchronized to light cues (Stokkan et al. 2001 ). This dissociation offers an exciting window through which new insights about the link between circadian disruption and metabolism can be explored. On the other hand, misalignment produced in mice by protocols resembling shift-work protocols may result in metabolic disturbances, such as the loss of glucose rhythmicity, inverted triglyceride rhythm, and an increase in body weight (Yoon et al. 2012).
Besides the timing of feeding, hyperlipidic diets also have an impact on the clock outputs, such as the free running period, amplitude of circadian clock protein oscillations, and phase-shift responses to light (Kohsaka et al. 2007; Mendoza et al. 2011). High-fat diets consumed ad libitum may affect the clock genes, reducing protein transcript levels, hormone receptors that regulate the clock, and clock-controlled genes involved in fuel utilization in the hypothalamus and peripheral tissues (Mendoza et al. 2008, 2011), also affecting the phase relationship between central and peripheral circadian oscillators (Pendergast et al. 2013).
Obese mice often show a less organized circadian activity, with bursts of locomotion more often observed during the rest phase of the cycle, more fragmented sleep, and a reduced amplitude of activity during the night (Laposky et al. 2009; Carmona-Alcocer et al. 2012; Fuentes-Granados et al. 2012). The way in which obesity affects circadian rhythms may also differ between sexes; obese female mice present more acute changes of circadian parameters than those observed in males, in an animal model where obesity occurs in an environment with free access to food but with limited space (Carmona-Alcocer et al. 2012).
The understanding of the mechanisms by which obesity is linked to the circadian clock is in its early stages. Different levels of organization in circadian regulation and metabolism can be observed. At the level of hypothalamic center that regulates feeding, the SCN projects axons to the arcuate nucleus, where the main integration of feeding–fasting cycles takes place (Green et al. 2008). Two main hormones, Ghrelin and Leptin, participate in feeding–fasting behavior through hypothalamic mediators. Ghrelin is an orexigenic hormone from the periphery (released mainly by the oxyntic cells within the stomach) that stimulates food intake and is elevated under conditions of physiological demand such as fasting (Schellekens et al. 2012). Leptin is mainly secreted by adipose tissue, and it is recognized as an endocrine signal related to the satiety process. It signals pro-opiomelanocortin (POMC) neurons in hypothalamus (De Jonghe et al. 2012). Both leptin and ghrelin also exert feedback on the SCN and are endocrine signals that are out of balance in conditions of obesity (Froy 2010).
At the cell level, chemical signals that couple internal clock function to nutrient state involve AMP kinase (AMPK). Stimulation of AMPK leads to degradation of CRY (Lamia et al. 2009), suggesting that AMPK is a key sensor and integrator of hormonal and nutritional signals with neurochemical and neurophysiological responses (Schneeberger and Claret 2012) that affect circadian transcriptional cycles. Glucocorticoids have also been shown to modulate the expression of lipogenic genes (Cho et al. 2012) and REV-ERB-α (Torra et al. 2000), a core clock gene involved together with PPARs in lipolytic metabolic pathways. NAD+ oscillation, redox flux, ATP availability, and mitochondrial function can also participate in tuning the clock by posttranslational modification of transcription factors (Bass 2012). NAD+/NADH+H oscillation regulates a protein deacetylase, Sirtuin 1, recently recognized as a key metabolic sensor in several tissues; it modulates a diversity of cellular processes, including glucose and lipid metabolism, fat mobilization, insulin secretion, sensing of nutrient availability in the hypothalamus, and the activity of the circadian clock in metabolic tissues (Li 2013).
Increasing research leads us to conclude that the relationships between circadian clock genes, central and peripheral oscillators as well as inputs and outputs to the SCN, represent a multifactorial system that must eventually be taken into consideration for treatments of obesity.
In humans, the meal timing as well as the food quality and quantity may be considered key factors contributing to metabolic impairment, seen as circadian disruption from consuming calories during the “wrong” circadian time. Circadian misalignment, such as occurs with jet lag or chronically with shift work, increases postprandial glucose, insulin, and mean arterial pressure, while it decreases leptin and sleep efficiency and completely inverts the cortisol profile across the behavioral cycle (Scheer et al. 2009).
Better planning and control of our environment (sleep cycles, time of eating and exercise, light exposure, etc.) will allow us to reduce the chances of circadian misalignment and may also help reduce the severity of various other diseases (Mikhail 2009).
14.8 Chronobiology of Cancer
Cancer is a group of diseases characterized by chronic proliferation of cells having many competetive advantages compared with normal cells. Two models have been proposed to explain the heterogeneous potential of tumor cells and the process of metastasis: (1) the stochastic model, in which a distinct population of cells acquires some somatic mutations and develops metastatic capability, and (2) the hierarchical model, in which primary tumors and metastatic cancer are initiated by occasional stem cells with cancerous properties (Reya et al. 2001; Li et al. 2007).
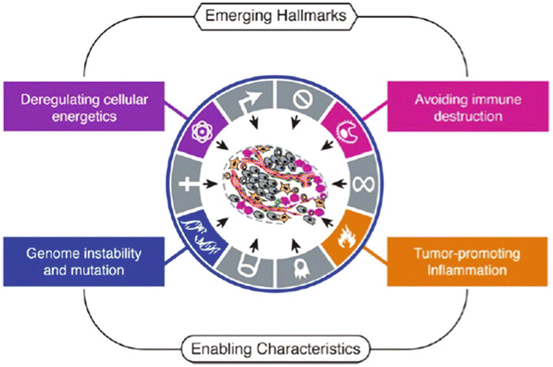
Hanahan and Weinberg in 2000 proposed to characterize the cancerous process by six hallmarks that constitute a logical framework for understanding the remarkable diversity of neoplasic diseases (Fig. 14.3).
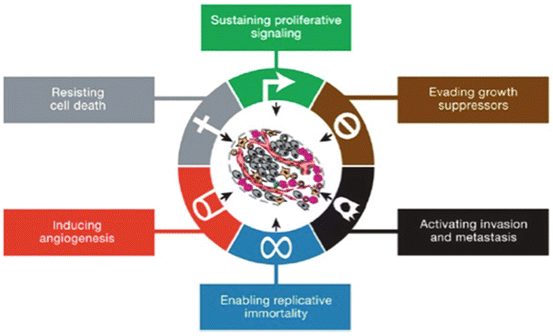
Fig. 14.3
Six hallmark capabilities proposed by Hanahan and Weinberg in 2000 to understand the multistep process of transformation from a healthy cell to neoplasic cell
14.8.1 Proliferative Signaling
The most fundamental characteristic of cancer cells is their ability to maintain chronic proliferation. Normal tissues control the release of growth-promoting signals that direct the progression through the cell cycle, therefore ensuring the correct tissue architecture and function. Cancer cells send signals to stimulate normal cells to support the tumor-associated stroma (Cheng et al. 2008; Bhowmick et al. 2004). Receptor signaling can also be deregulated by elevating the levels of receptor proteins displayed at the cancer cell surface, rendering such cells hyperresponsive to otherwise limiting amounts of growth factor ligands.
14.8.2 Evading Growth Suppressors
Cancer cells must also circumvent powerful programs that downregulate cell proliferation, mainly by the actions of tumor suppressor genes. Dozens of these genes have been discovered through their effects in one or another type of animal or human cancer; many of these genes have been validated by gain- or loss-of-function experiments in mice. Two prototypical tumor suppressors, RB (retinoblastoma-associated) and TP53 (tumoral p53) proteins, operate as central controls that govern the decisions of cells to proliferate or, alternatively, activate senescence and apoptotic programs (Hanahan and Weinberg 2011).
14.8.3 Resisting Cell Death
Experimental and clinical evidence has revealed that apoptosis is attenuated in tumors that progress to states of high-grade malignancy and resistance to therapy. The loss of TP53 tumor suppressor function is one of the principal factors associated with cellular immortalization. Alternatively, tumors may also increase the expression of anti-apoptotic regulators (Bcl-2, Bcl-xL) or of survival signals (IGF-1/2), or downregulate pro-apoptotic factors (Bax, Bim, Puma). The multiplicity of apoptosis-avoiding mechanisms presumably reflects the diversity of apoptosis-inducing signals that cancer cell populations adapt during their evolution to malignancy. The concept of “programmed cell death” has been broadened to include other forms of cellular death that are inhibited during the cancerous process (Hanahan and Weinberg 2011).
14.8.4 Enabling Replicative Immortality
Cancer cells require unlimited replicative potential in order to generate macroscopic tumors. This capability contrasts with the behavior of normal cell lineages, which are able to pass through only a limited number of successive cell growth-and-division cycles. This limitation has been associated with two distinct barriers to proliferation: senescence, a typically irreversible entrance into a non-proliferative but viable state, and crisis, which involves cell death. Accordingly, when cells are propagated in culture, repeated cycles of cell division lead first to the induction of senescence and then, for those cells that succeed in circumventing this barrier, to a crisis phase, in which the great majority of cells in the population die. On rare occasions, cells emerge from a population in crisis with unlimited replicative potential. This transition has been known as immortalization (Hanahan and Weinberg 2011). Multiple lines of evidence indicate that telomeres protecting the ends of chromosomes are centrally involved in the capacity for unlimited proliferation (Blasco 2005; Shay and Wright 2000).
14.8.5 Inducing Angiogenesis
Like normal tissues, tumors require nutrients and oxygen as well as the ability to evacuate metabolic waste and carbon dioxide. The tumor-associated new vasculature, generated by the process of angiogenesis, fulfills these needs. During tumor progression, an “angiogenic switch” is activated, causing quiescent vasculature to continually sprout new vessels that sustain expanding neoplastic growths (Hanahan and Folkman 1996).
14.8.6 Activating Invasion and Metastasis
As cancer develops and becomes metastasic, there are alterations in its shape as well as in its attachment to other cells and to the extracellular matrix (ECM). The best-characterized alteration involves the loss of E-cadherin, a key cell-to-cell adhesion molecule. By forming adherent junctions between epithelial cells, E-cadherin helps to maintain the quiescent state of the cells. Increased expression of E-cadherin is protective against cancer invasion and metastasis, whereas its reduction potentiates these phenotypes. The frequently observed downregulation and occasional mutational inactivation of E-cadherin in human carcinomas support its role as a key suppressor of metastasis (Berx and van Roy 2009; Cavallaro and Christofori 2004). Additionally, expression of genes encoding other cell-to-cell and cell-to-ECM adhesion molecules has been shown to be altered in some highly aggressive carcinomas.
In 2011, Hanahan and Weinberg added another two hallmarks to the cancerous process (Fig. 14.4).