CHAPTER 4 Pharmacologic Principles
DEFINITIONS IN PHARMACOKINETICS
Pharmacokinetic information is used to determine drug dosage regimens in clinical patients. An understanding of the way in which drug dosage regimens are derived and how they can be adjusted for different disease states requires knowledge of some basic pharmacokinetic terms. Mathematical models provide equations to describe drug concentration as a function of time. With an open model the drug is eliminated from the body. An open model describes the fate of most drugs. With a closed model the drug is recirculated within the body (e.g., a drug that undergoes enterohepatic recirculation). In pharmacokinetic models the body is represented by a series of compartments that communicate reversibly with one another. A compartment is a tissue or group of tissues with similar blood flow and drug affinity. A drug is assumed to be uniformly distributed within a compartment and can move dynamically in and out of compartments. Rate constants represent the entry and exit of drugs from each compartment. The central compartment is made up of the highly perfused tissues that equilibrate rapidly with the drug. Overall drug elimination occurs mainly from the central compartment, because the kidneys and liver are well-perfused tissues. The peripheral compartment is made up of less-perfused tissues such as muscle and connective tissues. The deep compartment consists of slowly perfused tissues or depot tissues such as fat and bone. The presence of a deep compartment for drug distribution is important for toxins and drug residues. Most drugs in clinical use are described by one or two compartment models. Models with more than three compartments are usually not physiologically relevant. Describing drug disposition with compartment models creates differential equations that describe drug concentration changes in each compartment and provides a visual representation of the rate processes among compartments.
RATES AND ORDERS OF REACTIONS
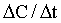
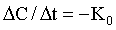
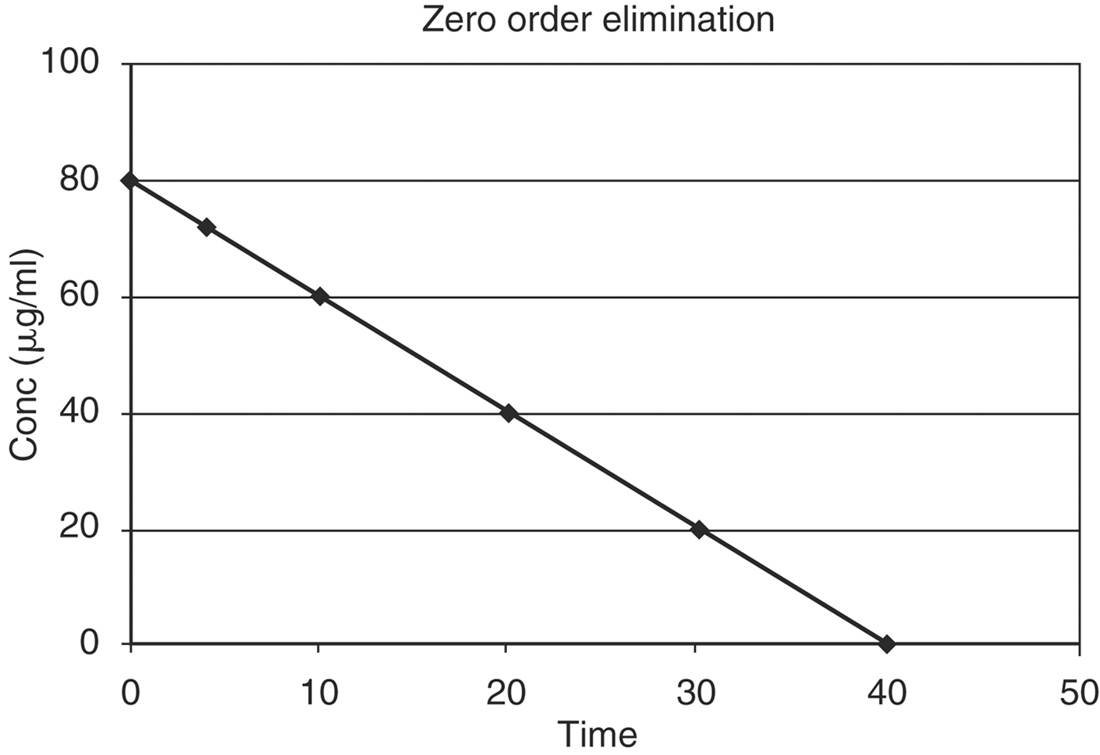
where K0 is the zero order rate constant in mg/ml min. A graph of drug concentration versus time on regular graph paper for a zero order reaction produces a straight line (Figure 4-1), described by the equation:
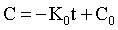
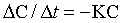
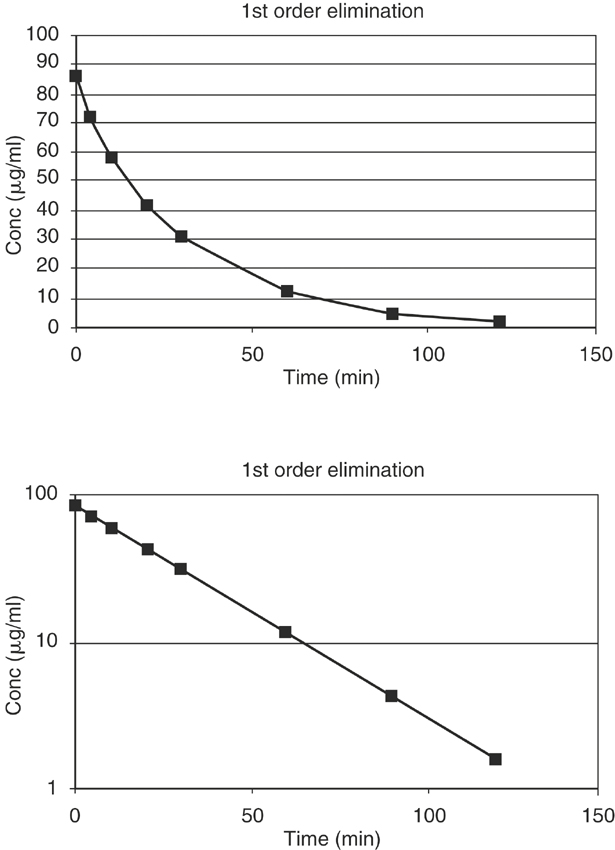
where K is the first order rate constant, is expressed in units of time-1 (min-1 or hr-1), and defines the fraction of drug eliminated from the body per unit time; C is the plasma drug concentration at any time t. Although K remains constant, the rate (>C/>t) is always changing because C is always decreasing. A graph of drug concentration versus time for a first order reaction produces an exponential curve on regular graph paper but produces a straight line on semilogrithmic graph paper (Figure 4-2) and is described by the following equation:
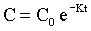
CLINICAL APPLICATION OF COMPARTMENTAL MODELING, RATES, AND ORDERS OF REACTIONS
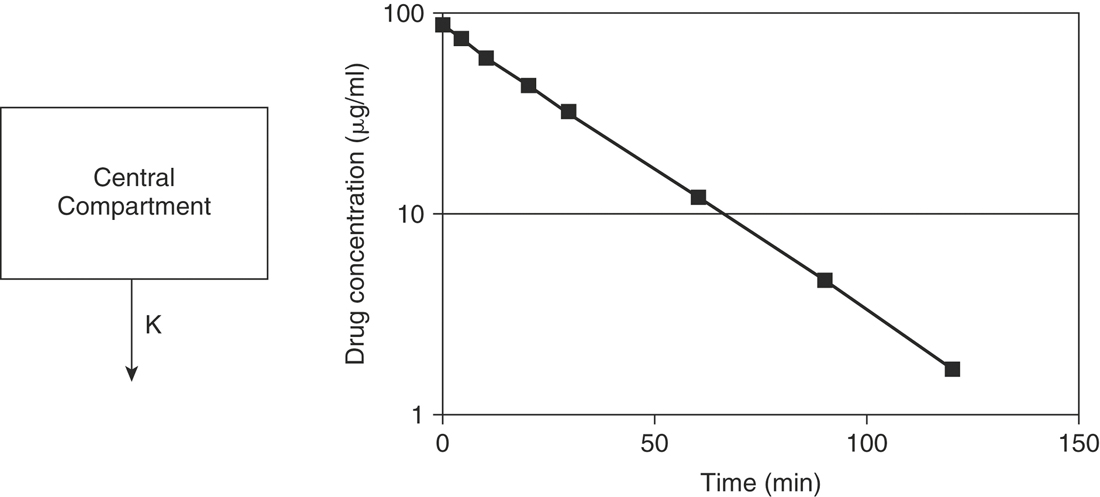
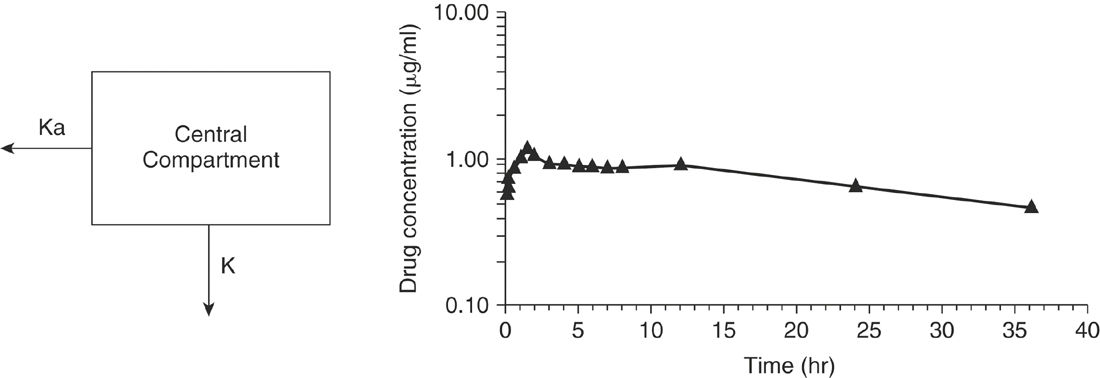
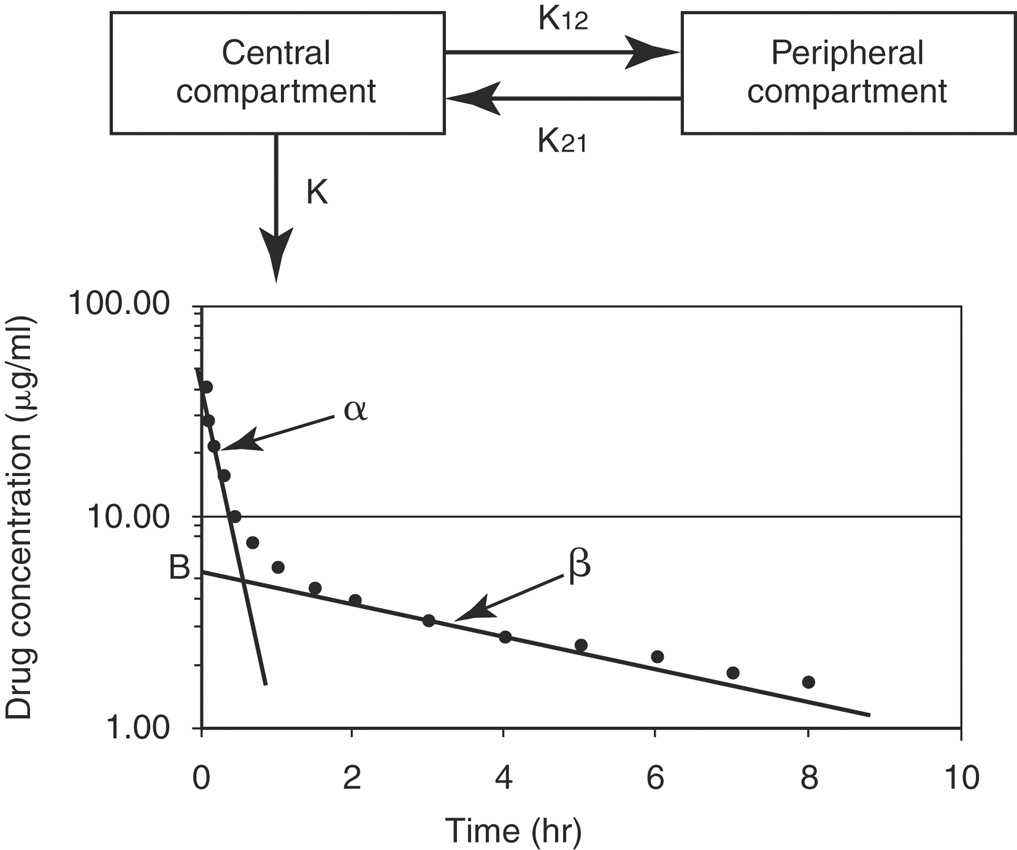
The aforementioned concepts can be combined to mathematically describe the changes in the drug concentration in the body over time. Drug disposition described by a one-compartment open model with intravenous (IV) injection and first order elimination (Figure 4-3) means that the body acts as one homogeneous compartment. A drug’s concentration in one part of the body is assumed to be proportional to its concentration in any other part. Many drugs administered by routes other than IV, such as oral, subcutaneous, intramuscular, or intradermal, are described by a one-compartment open model with first order absorption (Ka) and elimination (K) (Figure 4-4). With a two-compartment open model with IV injection and first order elimination, the model assumes the body acts as two compartments: the central compartment (blood and highly vascularized tissues) and a peripheral compartment (less vascularized tissues). Most drugs administered in veterinary medicine are described by this model (Figure 4-5). Elimination is considered to occur only from the central compartment, because the liver and kidneys are highly vascularized tissues. The plasma concentration versus time graph does not produce a straight line on semilogrithmic paper but can be broken into two sections and described by the following biexponential equation:
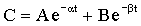
DISTRIBUTION OF DRUGS IN THE BODY
The volume of distribution (Vd) of a drug is the mathematical term used to describe the apparent volume of the body in which a drug is dissolved.1 The Vd is the parameter used to assess the amount of drug in the body from the measurement of a “snapshot” plasma concentration. The numerical value of Vd can give some indication of the distribution of the drug in the body. A drug’s distribution is determined by its ability to cross biologic membranes and reach tissues outside the vascular system. The physical characteristics of the drug molecule, such as ionization, lipid solubility, molecular size, and degree of protein binding, determine its ability to cross biologic membranes.
Three volumes of distribution (Vd) are reported in the veterinary literature: the volume of the central compartment, the steady-state volume of distribution, and the volume of distribution calculated by the area method. Conceptually, the easiest demonstration of the volume of distribution is with the volume of the central compartment (Vdc). Just after an IV dose, plasma drug concentration is maximal (Figure 4-6). Assuming that the instant drug concentration (C0) results from the drug mixing in the blood, the Vdc is the apparent volume from which drug elimination occurs, because the kidneys and liver belong to the central compartment, and is calculated from the following equation:
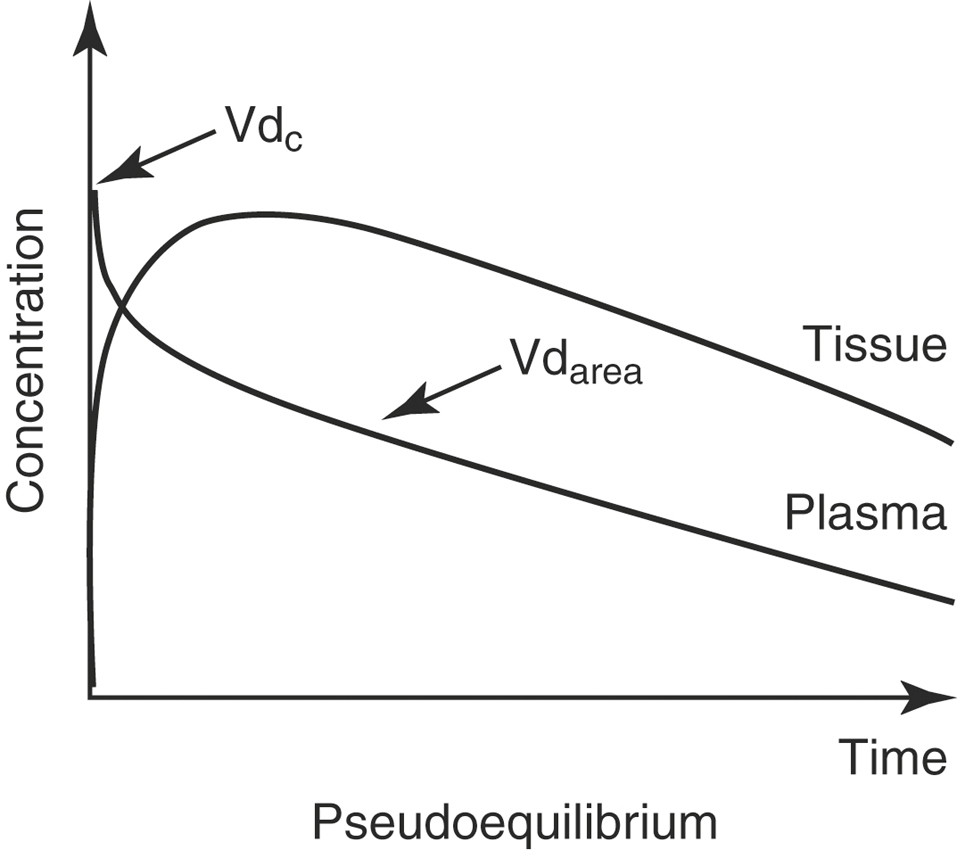
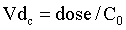
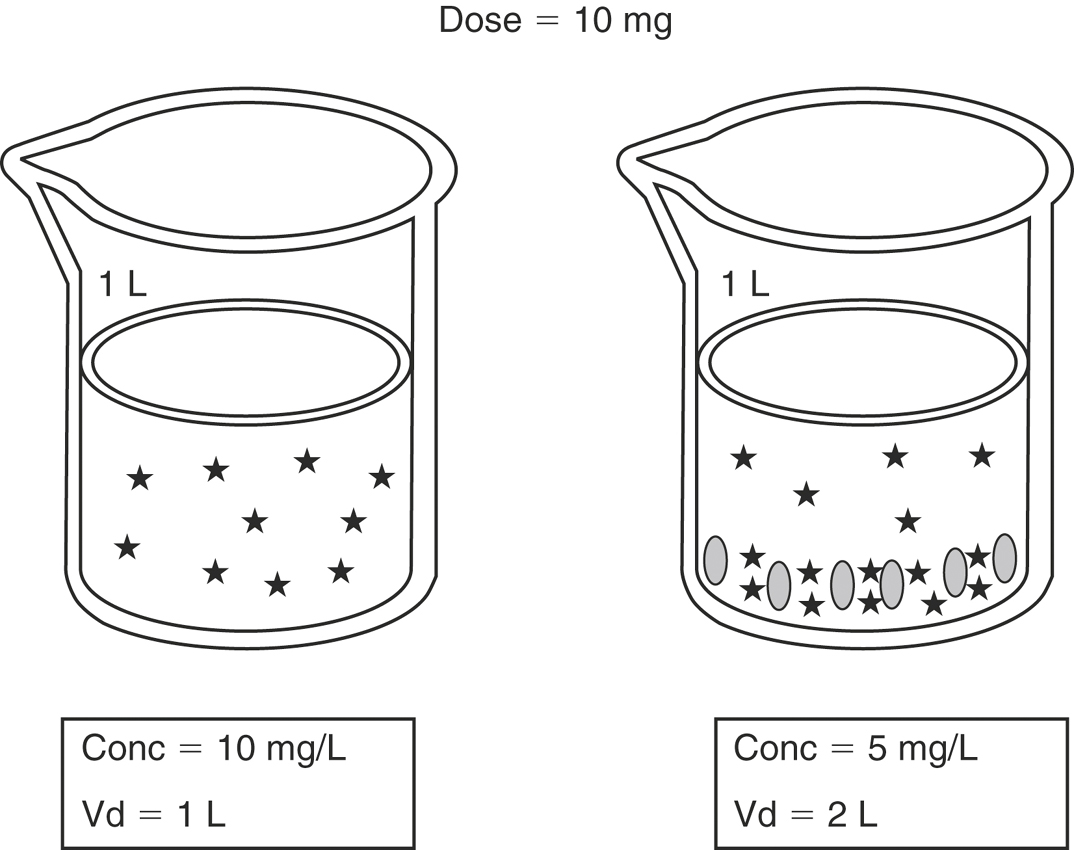
where C0 is the concentration at time zero, extrapolated from the plasma concentration versus time graph. To understand what the Vdc for a drug represents, consider the body as a beaker filled with fluid (Figure 4-7). The fluid represents the plasma and other components of extracellular water. If a drug is administered intravenously, it rapidly distributes in the extracellular fluid. If the drug does not readily cross lipid membranes, it will be confined mainly to the extracellular fluid and a plasma sample therefore will have a high drug concentration. The higher the measured concentration in relation to the original dose, the lower the numerical value for Vdc. Drugs such as the β-lactam and aminoglycoside antibiotics are poorly lipid soluble and therefore remain predominantly in the extracellular fluid and have low values for Vd. In contrast, some drugs readily cross lipid membranes and distribute into tissues. This is represented by the beaker on the right, where the stars at the bottom of the beaker represent drug molecules that have been taken up by tissues. A plasma sample will have a low drug concentration in proportion to the original dose and therefore will have a high numerical value for Vdc. Given the limitations on measuring drug concentrations at “time zero” and using the aforementioned formula, the measured concentration of highly lipid-soluble drugs can be low enough to result in a value of Vdc that is greater than 1 L/kg, so it is often referred to as an apparent volume of distribution. In the example on the right, the “apparent” Vd is 2 L/kg even though the beaker contains only 1 L of fluid.
For most drugs after a single IV dose, the drug is distributed and begins to be eliminated simultaneously. When concentrations are measured and the data graphed, there is a distribution phase, wherein the plasma drug concentration that is due to elimination and not distribution increases until it reaches an asymptotic value at which pseudoequilibrium is achieved (see Figure 4-6). When pseudoequilibrium is reached, the movement of drug between the peripheral and central compartments reaches equilibrium, and decreasing plasma concentrations are now due only to irreversible elimination (described by the elimination rate constant, β). The applicable Vd value in this situation is the volume of distribution by area (Vdarea),
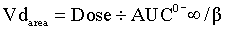
With an IV infusion or with a multiple dose regimen, the rate of drug entry into the body is equal to its elimination rate, and the body becomes a closed system with no clearance. In this situation the correct Vd to describe distribution is the Vd at steady-state (Vdss; Figure 4-8):
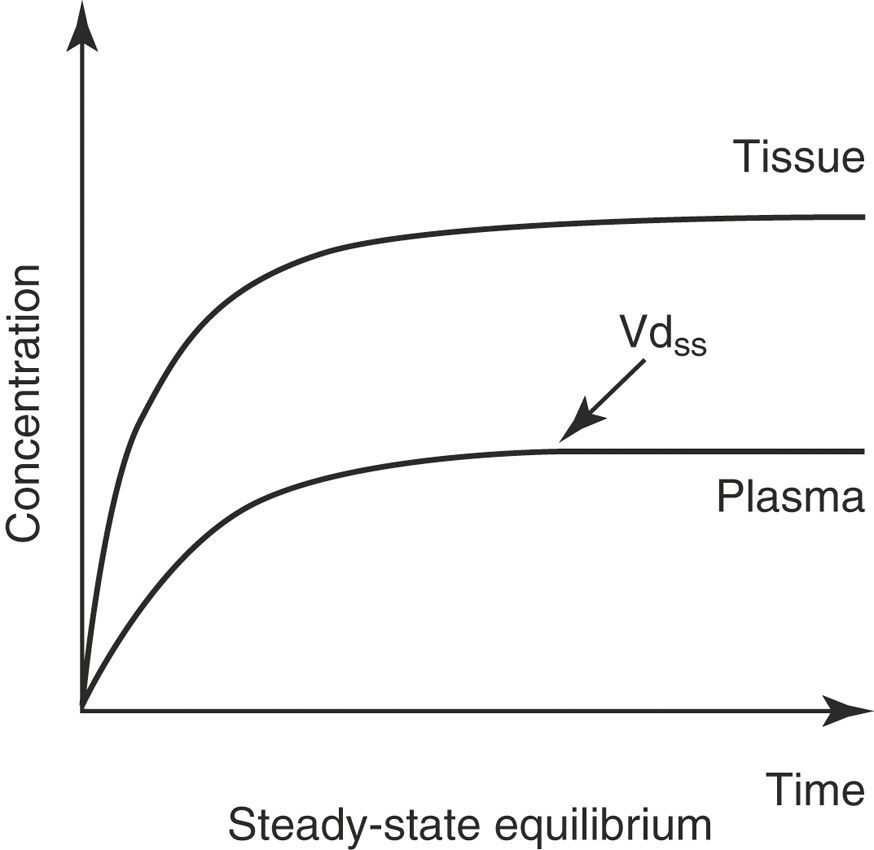
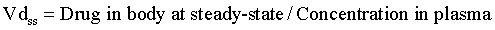
Clinical Use of the Different Volume of Distribution Values
It is useful to compare a drug’s Vd to the distribution of water in the body in order to get an idea of its distribution. Drugs with a Vd value of less than 0.3 L/kg are predominantly confined to the ECF, whereas drugs with a Vd value of greater than 1 L/kg are highly lipid soluble and tend to distribute out of the extracellular fluid and into tissue compartments (Box 4-1). Although the value of Vd does not confirm penetration of a drug into specific tissues, in general the higher the value of the Vd, the more likely it is that the drug will reach sequestered sites such as the brain and cerebrospinal fluid, the prostate and other sex organs, the eye, and the mammary gland. Studies must be performed to confirm that therapeutic concentrations are achieved in such sites.
Conditions That Affect Volume of Distribution
The Vd is constant for any drug and will change only with physiologic or pathologic conditions that change the distribution of the drug. Drugs with high Vds are usually very lipid soluble and typically are not significantly affected by changes in body water status and do not require dosage adjustment. However, there are many medical conditions that affect the disposition of low Vd drugs in a patient (e.g., nonsteroidal anti-inflammatory drugs and aminoglycosides), and these drugs do require dosage adjustment because of their narrow therapeutic index. Many conditions in horses, such as colic, are characterized by volume contraction and dehydration and changes in acid-base balance, which affect the extracellular fluid volume. Neonatal foals have a higher percentage of body water than adult horses (80% versus 60% total body water), and the extra 20% is primarily confined to the extracellular fluid, so the Vd of drugs such as gentamicin are higher in neonatal foals than older foals or adult horses.2 Therefore to achieve equivalent therapeutic plasma concentrations of gentamicin in a neonatal foal, the dose must be higher than that administered to the older foal or adult horse.
Bioavailability
Bioavailability (F) is a measure of the systemic availability of a drug administered by a route other than IV.3 Bioavailability is determined by comparing the area under the plasma drug concentration curve versus time (AUC) for the extravascular formulation to the AUC for the IV formulation. The AUC is calculated by computer or by the trapezoidal method, wherein the entire curve is divided into trapezoids, then the area of each trapezoid is calculated and summed to give the AUC. For an orally administered drug, use the following equation:
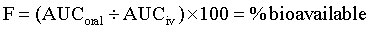
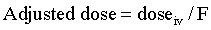
If the oral formulation of a drug has a mean bioavailability of 50%, the drug dose must be doubled to achieve the same concentrations in plasma as achieved using the IV formulation. However, the variability of the bioavailability in the population is more clinically significant than the mean. To make sure that the horse with the poorest absorption is dosed appropriately, the dose must be increased according to the lowest bioavailability, not the mean. For example, if a drug has a mean F of 50% with a range of 20% to 70%, then to achieve an exposure of 100% for all the treated horses, the dose must be multiplied by 5, not just 2. However, if this is done, the horses with an F of 70% will be overdosed by a factor of 3.5. For a drug with a narrow therapeutic window and a poor bioavailability, there may be no dose that is ideal for all horses in the population. Low bioavailability of antimicrobials and anthelmintics is a major cause of subtherapeutic dosages that promote drug resistance. Poor oral bioavailability is a major limitation of many drugs administered to horses.4
Lipid Solubility and Drug Ionization (the pH-Partition Hypothesis)
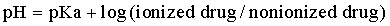
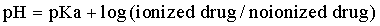
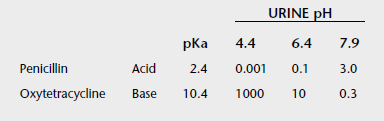
Whereas the precise ratios of ionized versus nonionized drug can be calculated from the Henderson-Hasselbach equations, the relevance of the equations can be understood by simply remembering the sentence “like is nonionized in like.” For example, a weak acid will be most nonionized in an acidic environment, so aspirin is most nonionized in the stomach and is readily absorbed. The fluid of most sequestered sites in the body (cerebrospinal fluid, accessory sex gland fluid, milk, abscesses) has a pH more acidic than plasma. In cattle with mastitis weak acid antibiotics are typically administered by intramammary infusion, whereas weak bases are administered parenterally. This makes sense according to the pH-partition concept. Weak bases in the plasma are highly nonionized and readily cross into the mammary gland. Then, as the equilibrium shifts, they become “ion-trapped” in the more acidic milk, but the fraction of nonionized drug in the mammary gland is available to cross the bacterial cell membrane for antimicrobial action. Weak acids such as penicillins and cephalosporins are highly ionized in plasma and therefore do not penetrate into the mammary gland very well, so these are most effective when administered by local infusion into the udder, where the extremely high local concentrations negate local pH effects.
Typically, drugs that are weak acids will have low Vd values and weak bases will have high values for Vd (Box 4-2). Amphoteric drugs such as the fluoroquinolones and tetracyclines have acidic and basic groups on their chemical structures. These drugs have a pH range where they are maximally nonionized. For example, enrofloxacin is most lipid soluble (nonionized) in the pH range of 6 to 8, so it is lipid soluble at most physiologic pHs. In acidic urine significant ionization occurs, which reduces enrofloxacin’s antibacterial activity. But this reduction in activity is offset by the extremely high concentrations of enrofloxacin achieved in urine, so it is of no clinical importance. Despite being weak bases, the aminoglycosides are very large, hydrophilic molecules and have high pKa values, so they are highly ionized at physiologic pHs. Therefore parenterally administered aminoglycosides do not cross lipid membranes well and do not achieve therapeutic concentrations in milk, accessory sex gland fluids, abscesses, or cerebrospinal fluid.
Drug Protein Binding
Protein binding can involve plasma proteins, extracellular tissue proteins, or intracellular tissue proteins. Many drugs in circulation are bound to plasma proteins, and because bound drug is too large to pass through biologic membranes, only free drug is available for delivery to the tissues and to produce the desired pharmacologic action. Therefore the degree of protein binding can greatly affect the pharmacokinetics of drugs. Acidic drugs such as nonsteroidal anti-inflammatory drugs tend to bind predominantly to albumin.5 Albumin is the most abundant plasma protein, and it is critical to maintaining the colloidal oncotic pressure in the vascular system. As a negative acute phase protein, albumin concentration decreases during inflammation. Hypoalbuminemia results from decreased production, seen with severe hepatic insufficiency, or by loss through increased rates of urinary excretion, such as in nephrotic syndrome or with mucosal damage, as with protein losing enteropathies. Basic drugs typically bind to α-1 acid glycoprotein, which is an acute phase protein, whose hepatic production increases significantly with inflammatory conditions.6 Other proteins, including corticosteroid binding globulin, are important for binding of some specific drugs but are less important in overall drug-protein binding.7 There is equilibrium between free and bound drug, however, just like the relationship of ionized and nonionized drug molecules. Protein binding is most clinically significant for antimicrobial therapy, where a high degree of protein binding serves as a drug “depot,” allowing for increased duration of the time the drug concentration remains above the bacterial minimum inhibitory concentration, adding to antimicrobial efficacy.8 For other drugs changes in plasma protein binding can influence individual pharmacokinetic parameters, but changes in plasma protein binding usually do not influence the clinical exposure of the patient to a drug. Changes in protein binding caused by drug interactions are assumed to instantaneously change free drug concentrations and have been frequently cited as a cause of adverse drug reactions. But the increase in free drug concentration is only transient, because drug distribution and drug elimination change to compensate. The often-cited example of the concurrent administration of phenylbutazone and warfarin leading to bleeding caused by increased free concentrations of warfarin is erroneous. The true interaction is from phenylbutazone-induced inhibition of the hepatic metabolism of warfarin, which results in increased plasma concentrations and increased anticoagulant effect.7 Therefore adjustments in dosing regimens because of hypoproteinemia or concurrent administration of highly bound drugs are not necessary except in the rare case of a drug with a high hepatic extraction ratio and narrow therapeutic index that is given parenterally (e.g., IV dosing of lidocaine).9
ELIMINATION RATE CONSTANT AND ELIMINATION HALF-LIFE
The rate of elimination for most drugs is a first order process. The elimination rate constant (K) represents the sum of drug elimination by excretion and metabolism. Drug elimination is considered always to occur from the central compartment, because the liver and kidney are well-perfused tissues. The elimination rate constant is used to calculate the drug’s half-life (T½), or the time required for drug concentration to decrease by one half (Figure 4-9). For first order reactions, T½ is constant across the plasma concentration versus time curve and is calculated from:
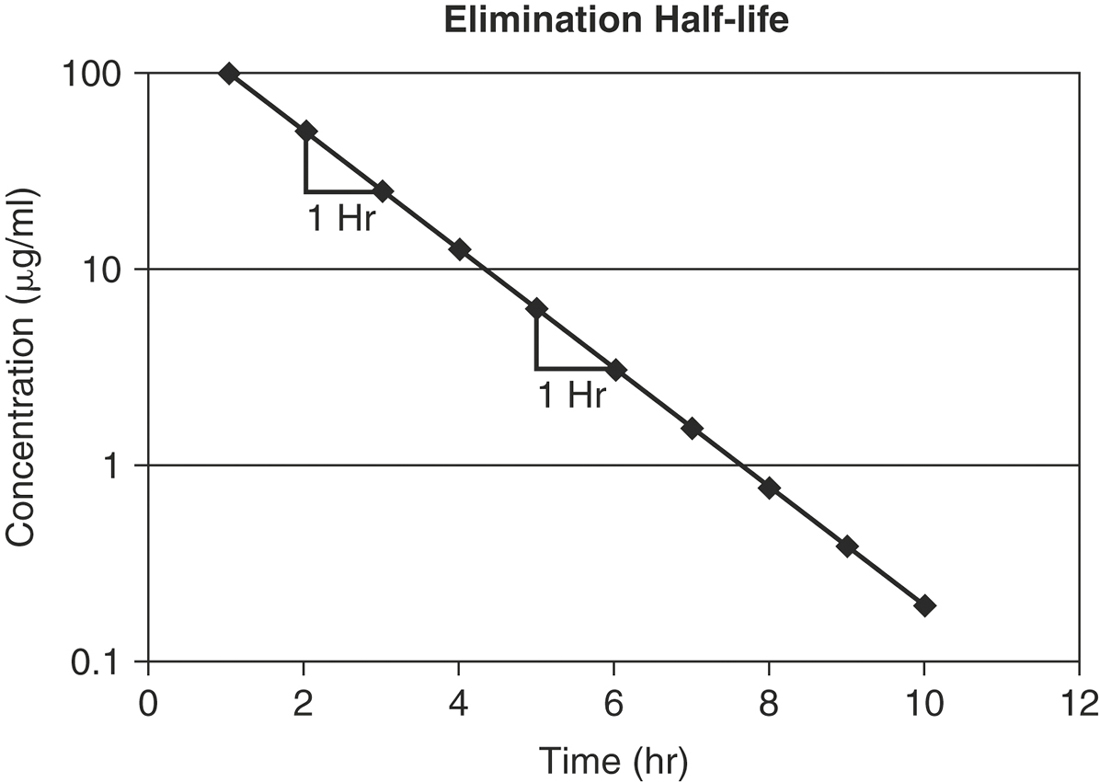
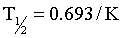
where 0.693 = ln2 (the natural logarithm of 2). Mean residence time (MRT) is roughly the equivalent of T½ when pharmacokinetics are calculated using statistical moment theory. The MRT is the time it takes for drug concentration to decrease by 63.2%, so the MRT value is typically slightly greater than T½. The T½ determines the drug dosage interval, how long a toxic or pharmacologic effect will persist, and drug withdrawal times for food animals or performance horses. Notice that it takes 10 T½s to decrease the plasma concentration by 99.9% (Table 4-1). Knowing a drug’s plasma T½ can give the clinician some idea of the drug’s withdrawal time for food or performance animals. However, for drugs that undergo hepatic metabolism (e.g., phenylbutazine) or drugs that sequester in specific tissues (e.g., aminoglycosides, isoxuprine), simply multiplying the T½ by a factor of 10 for a withdrawal time may not be sufficient to prevent violative residues. Also note that doubling a drug dose does not double the withdrawal time; it merely adds one half-life to the time it takes to reach the acceptable threshold concentration (Figure 4-10).
TABLE 4-1 Half-Life of Elimination of a Drug
| Number of Half-Lives | Fraction of Drug Remaining (%) |
|---|---|
| 0 | 100 |
| 1 | 50 |
| 2 | 25 |
| 3 | 12.5 |
| 4 | 6.25 |
| 5 | 3.125 |
| 6 | 1.56 |
| 7 | 0.78 |
| 8 | 0.39 |
| 9 | 0.195 |
| 10 | 0.0975 |
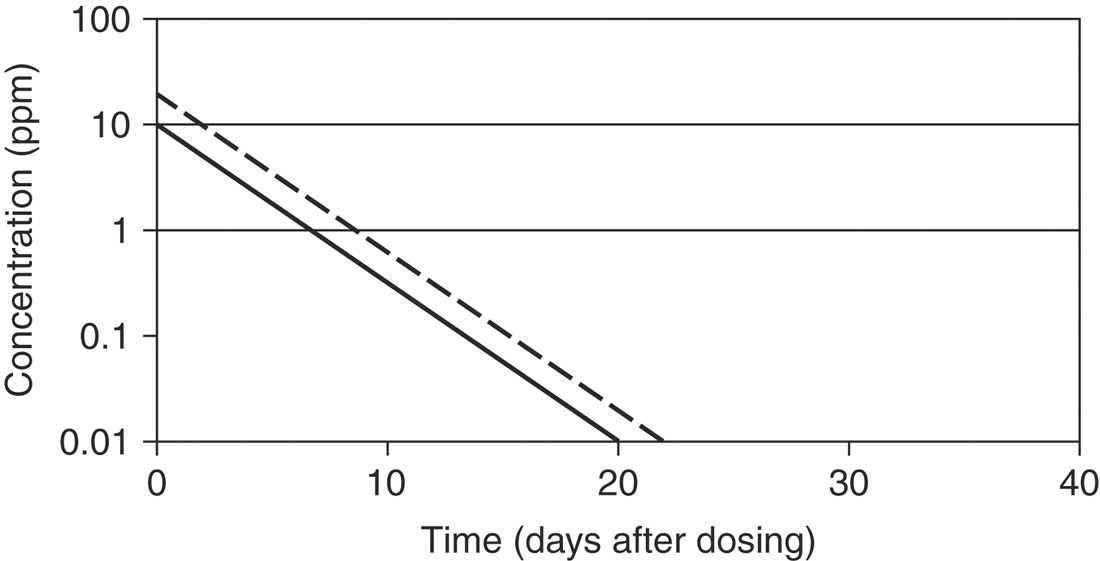
Flip-Flop Kinetics
Long-acting drug formulations are often products whose carriers cause them to be slowly absorbed into the systemic circulation. Therefore the drug elimination rate is limited by the drug absorption rate. The value for K (the elimination rate constant) calculated from the plasma concentration versus time curve is actually the value for Ka (the absorption rate constant). The easiest way to identify “flip-flop” kinetics is to compare the plasma concentration versus time curve for the extravascular route of administration to the curve after the drug is given intravenously (Figure 4-11). If the elimination phases of the curves are not parallel, then delayed absorption is prolonging elimination and the flip-flop phenomenon has occurred.
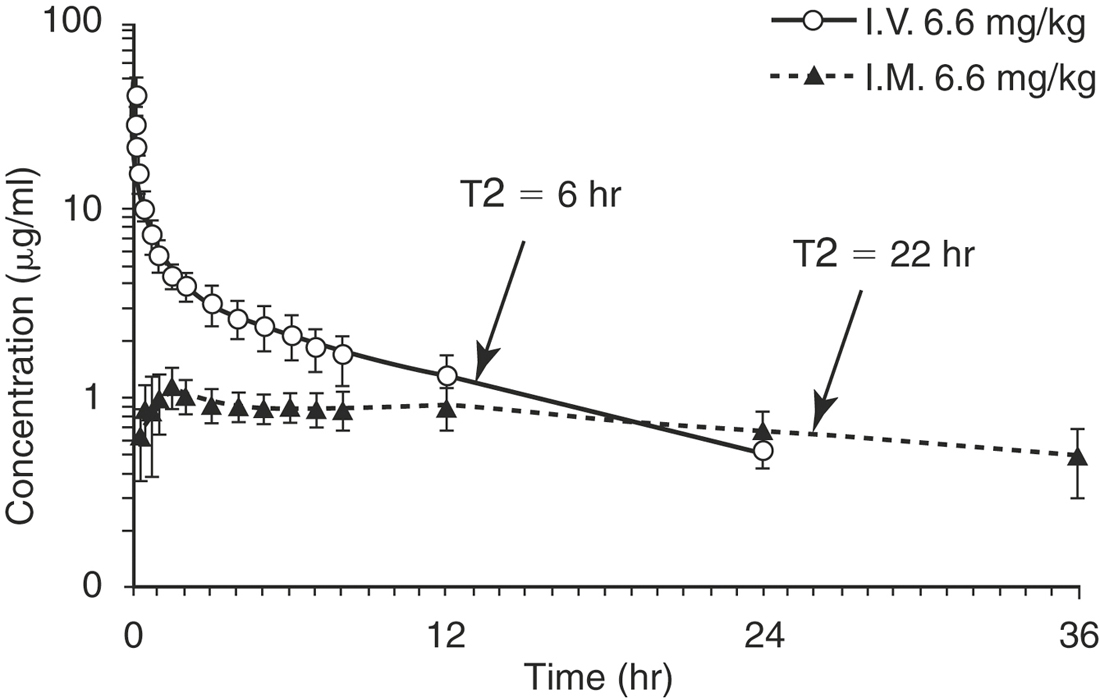
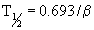
Clearance
Clearance is a measure of drug elimination from the body without reference to the mechanism of elimination. It is always reported in pharmacokinetic papers, but its significance is rarely explained in pharmacokinetic studies in horses. Clearance is the most important pharmacokinetic parameter because it is the only parameter that controls overall drug exposure and it is used to calculate the dosage required to maintain a specific average steady-state concentration.10
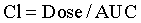
where AUC is the area under the plasma concentration time curve.
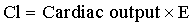
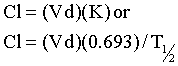
Consider the values for clearance and T½ values for four antimicrobial drugs (Table 4-2). Note that the plasma clearance values are similar, but the elimination half-lives are very different. Because the T½ is influenced by the extent of drug distribution, the drugs have similar clearance, but oxytetracycline has the largest Vd and the longest T½. Because T½ is derived from rate constants and does not have a physiologic basis, it is influenced by the sensitivity of the analytical method and by many pharmacokinetic parameters, and it is a poor parameter alone to evaluate physiologic (e.g., age, sex) or pathologic (e.g., renal failure) changes that effect drug disposition.

Renal Clearance of Drugs
Renal excretion is the major route of elimination from the body for most drugs. Drug disposition by the kidneys includes glomerular filtration, active tubular secretion, and tubular reabsorption (Figure 4-12), such that renal drug clearance is defined by the following equation:
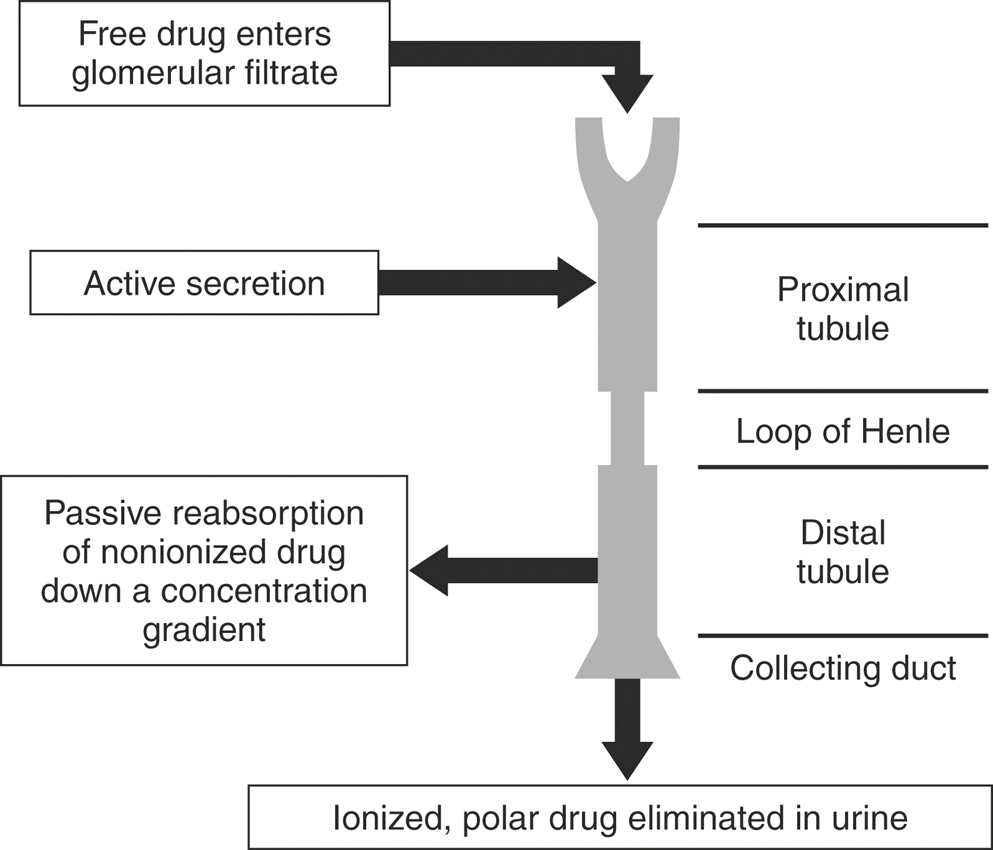
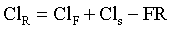
If ClR is greater than ClF, then some tubular secretion is occurring. Active tubular secretion is a carrier-mediated transport system, located in the proximal renal tubule. It requires energy input because the drug is moved against a concentration gradient. Two active tubular secretion systems have been identified: anion secretion for acids and cation secretion for bases. Drugs with similar structures may compete with each other for the same transport system. For example, probenecid competes with penicillin or the fluoroquinolones for the same transport system, effectively decreasing Cl of these antimicrobials. In patients with reduced functional renal tissue, remaining transport systems become easily saturated and drug accumulation occurs.
If ClR is less than GFR, tubular reabsorption of drug is occurring. Tubular reabsorption is an active process for endogenous compounds (e.g., vitamins, electrolytes, glucose). It is a passive process for the majority of drugs. It occurs along the entire nephron but primarily in the distal renal tubule. Factors that affect reabsorption include the pKa of the drug, urine pH, lipid solubility, drug size, and urine flow. Drug reabsorption is highly dependent on ionization, which is determined by the drug’s pKa and the pH of the urine. According to the Henderson-Hasselbach equation, a drug that is a weak base will be mainly nonionized in alkaline urine and a weak acid will be mainly ionized in alkaline urine. The nonionized form of the drug is more lipid soluble and has greater reabsorption (Table 4-3). The pKa of a drug is constant, but urinary pH is highly variable in animals and varies with the diet, drug intake, time of day, and systemic acidosis/alkalosis. Species differences have a major influence on the renal excretion of ionized drugs. Carnivores, with a urine pH of 5.5 to 7.0, will have a greater renal excretion of basic drugs than herbivores, with a urine pH of 7.0 to 8.0.
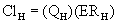
Biotransformation (Hepatic Metabolism) of Drugs
Metabolism is necessary for removal of lipophilic drugs from the body (Figure 4-13). Biotransformation depends on the chemical composition of the liver, activity of major drug metabolism enzymes, hepatic volume (perfusion rate), drug accessibility to and extraction by hepatic metabolic sites, and physicochemical properties of the drug. Biotransformation of a parent drug results in metabolites that may be active or inactive themselves. A prodrug is a drug administered in an inactive form that must be biotransformed to its active form, such as prednisone to prednisolone. Drug metabolic pathways are divided into phase I and phase II reactions. Phase I reactions (oxidation, reduction, hydrolysis, hydration, dethioacetylation, isomerization) typically add functional groups to the drug molecule necessary for phase II reactions. Phase II reactions (glucuronidation, glucosidation, sulfation, methylation, acetylation, amino acid conjugation, glutathione conjugation, and fatty acid conjugation) typically include conjugation reactions that increase the water solubility of the drug, facilitating excretion from the body.
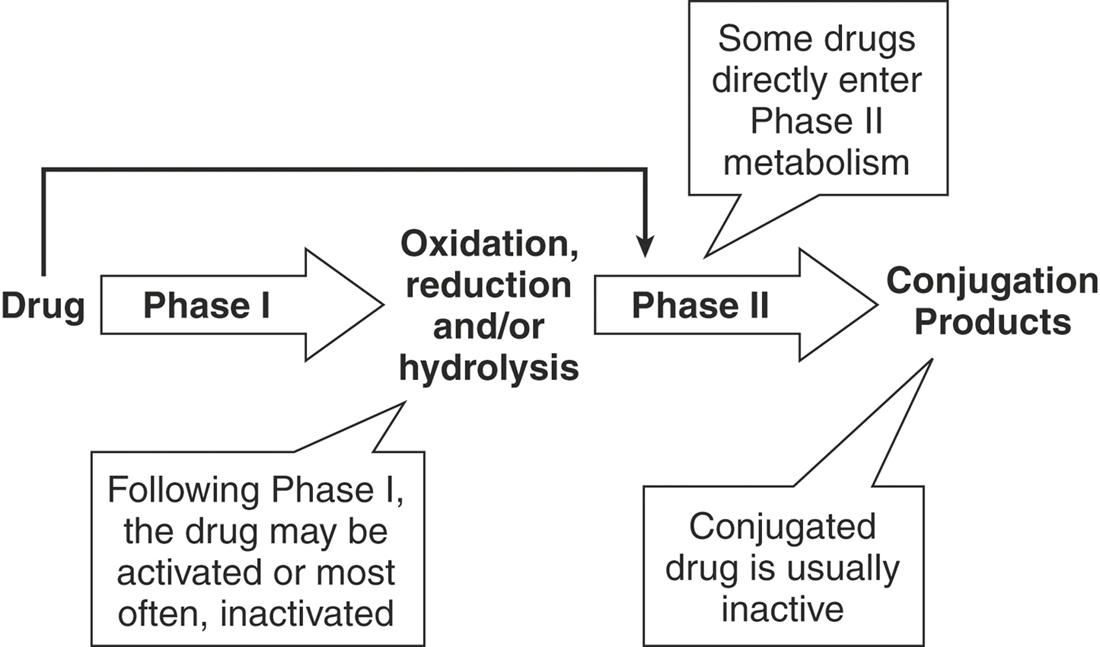
FIGURE 4-13 Hepatic metabolism increases the water solubility of drugs, facilitating excretion from the body.
Induction and Inhibition of Metabolism
Metabolism of drugs can be substantially affected by enzyme induction or inhibition by other drugs or chemicals (Box 4-3). In some cases the drug itself may alter its own metabolic fate by induction or inhibition. Many drugs are capable of inducing enzyme activity, thereby increasing the rate of metabolism and hepatic clearance of concurrently administered drugs, which typically results in a decreased pharmacologic effect. Enzyme induction typically occurs slowly, requiring several weeks to reach maximum effect. Induction is accompanied by increased hepatic ribonucleic acid (RNA) and protein synthesis and increased hepatic weight. Enzyme induction is important in the pathogenesis of hepatotoxicity and therapeutic failure of many drugs. Phenobarbital is a potent enzyme inducer known for hepatotoxicity and for inducing its own metabolism. Rifampin induces the metabolism of azole antifungals; concurrent administration with itraconazole results in subtherapeutic itraconazole concentrations.
Drug-induced enzyme inhibition also occurs and typically results in prolonged clearance of a concurrently administered drug. The potential for toxicity or an exaggerated pharmacologic response is increased. In contrast to induction, inhibition occurs rapidly. Erythromycin and enrofloxacin are known inhibitors of the metabolism of theophylline; concurrent administration can cause central nervous system toxicity and seizures.11,12
DRUG ACCUMULATION
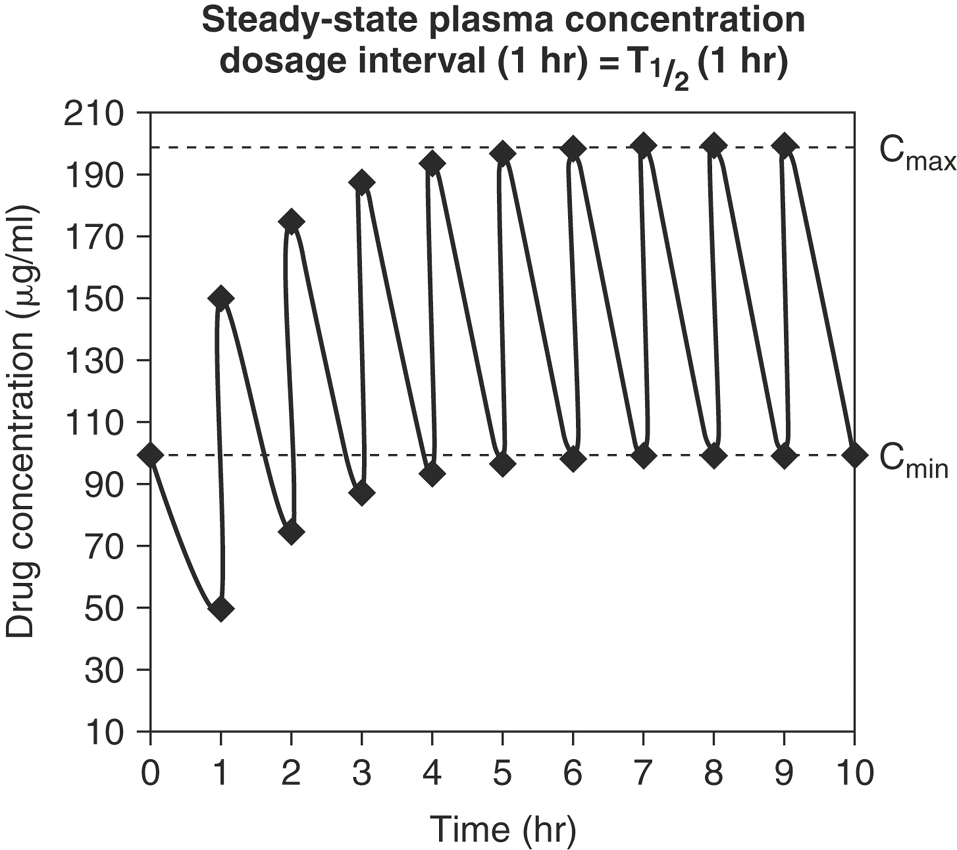
Drugs are often given in multiple-dosage regimens. To predict plasma drug concentrations, it is necessary to decide whether successive doses of a drug have any effect on the previous dose. The principle of superposition assumes that early doses of drug do not affect the pharmacokinetics of subsequent doses. For most drugs, as equal doses are given at a constant dosage interval, the plasma concentration-time curve plateaus and a steady-state is reached. At steady-state the plasma drug concentration fluctuates between a maximum concentration (Cmax, or peak) and minimum concentration (Cmin, or trough). Once steady-state is reached, Cmax and Cmin are constant and remain unchanged from dose to dose (Figure 4-14). The time to steady-state depends solely on the elimination half-life. It takes approximately six T½s to reach 99% steady-state levels. The drug dose and dosage frequency influence the values of Cmax and Cmin at steady-state, while the dosage frequency and T½ influence the fluctuation between Cmax and Cmin.
Clinical Consequences of Dosage Intervals Less than the Half-Life
Drugs such as phenobarbital, potassium bromide, phenylbutazone, and digoxin are commonly given at dosage intervals much shorter than their T½=s (Figure 4-15). This results in a Cmax at steady-state that is greater than the peak concentration after a single dose. There is minimal fluctuation between Cmax and Cmin, and missing a single dose will not affect plasma concentrations greatly. There is a lag time to reach the desired plasma concentrations at steady-state, and there will be a lag time for plasma concentrations to change in response to a dose change.
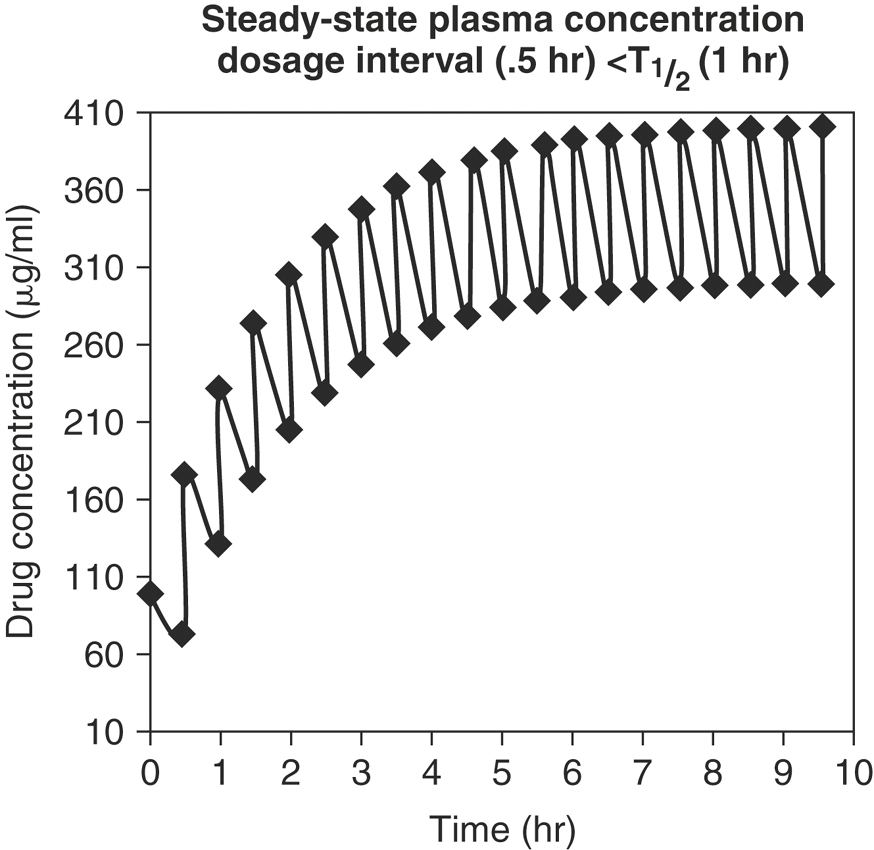
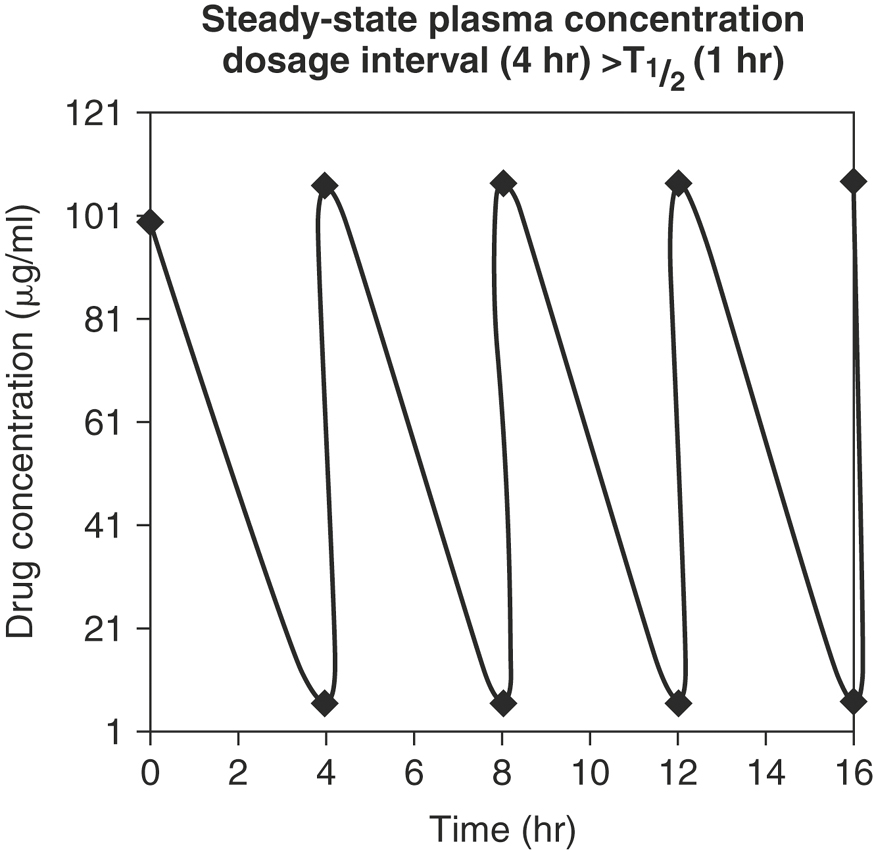
Clinical Consequences of Dosage Intervals Greater than the Half-Life
Drugs such as IV formulations of penicillin and cephalosporins are administered at dosage intervals that are greater than the T½ (Figure 4-16). As the dosage interval increases, Cmax at steady-state is closer in value to the peak concentration after a single dose. If the dosage interval is greater than 10 T½ s (the time required to eliminate 99.9% of the previous dose), drug accumulation essentially does not occur. There is marked fluctuation between Cmax and Cmin (peak and trough), and missing a dose will greatly affect plasma concentrations. However, there is minimal lag time to achieve the desired plasma concentration.
DESIGNING DRUG DOSAGE REGIMENS
The horse’s age may have a profound effect on drug disposition (Table 4-4).13 The definition of geriatric varies between species, and in small animals it varies between breeds. Body composition and regional blood flow change in geriatric horses. Cardiac output decreases, so regional and organ blood flow also decrease. These changes have an impact on drug absorption, distribution, and elimination. Blood flow is preferably redistributed to the brain and heart, so there is an increase in risk of drug toxicity in these organs. Gastrointestinal motility and absorptive capacity are reduced. Hepatocyte number and function decrease along with hepatic and splanchnic blood flow. As renal blood flow decreases, GFR and active secretory capacity of the nephron decreases, resulting in decreased renal clearance of drugs. Lean body mass decreases while fatty tissues increase. The plasma concentrations of water-soluble (low volume of distribution) drugs tend to increase, whereas the plasma concentrations of lipid-soluble (high volume of distribution) drugs tend to decrease. Serum albumin decreases while gamma globulins increase, so total plasma protein concentrations essentially remain the same.
TABLE 4-4 Age-Related Changes in Geriatric and Pediatric Patients
| Body Part/Function Affected | Geriatric | Pediatric |
|---|---|---|
| Organ blood flow | Decreased | Increased |
| Total body water | Decreased | Greatly increased |
| Body fat | Increased | Decreased |
| Serum proteins | Decreased albumin | Decreased albumin |
| Increased globulins | ||
| Hepatic metabolism | Decreased | Greatly decreased |
The definition of neonate also varies with species and age, but all the determinants of drug disposition are altered as the foal matures.14 Blood flow to the heart and brain is greater and faster, making the foal more susceptible to drug-induced cardiotoxicity and neurotoxicity. Gastrointestinal absorption tends to be decreased as a result of decreased gastric emptying and decreased intestinal peristalsis. Absorption from intramuscular and subcutaneous sites changes as muscle mass and blood flow change. Neonates have less fat and greater total body water (primarily extracellular fluid) than adults. Therefore low Vd drugs (e.g., gentamicin, ketoprofen) distribute into a larger volume, making it necessary to increase the dose to avoid therapeutic failure. Because of low body fat stores, lipid-soluble drugs will have higher plasma concentrations in foals. For example, moxidectin is much more lipid soluble than ivermectin, so it is more easily overdosed in foals.15 Drug elimination by both hepatic metabolism and renal excretion is limited in neonates, so drug dosing intervals need to be increased for many drugs, such as aminoglycosides and nonsteroidal anti-inflammatory drugs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


