Chapter 1. Pathophysiology and Tumor Cell Growth
Barbara E. Kitchell and Nikolaos G. Dervisis
Intricate and complex pathways are involved in the development of a multicellular eukaryotic organism. Each organism originates as a single cell and progresses through rapid proliferation and differentiation, followed by steady state repair and repletion, and ultimately senescence. The genetic information for every part of the mature organism exists in the genome of each cell, yet the individual differentiated cell never expresses the majority of this genetic information. By studying aberrancies in the system, such as the development of neoplasia, mechanisms that control the process of gene activation and repression in health and disease are being elucidated. Consequently, our understanding of what constitutes a “cancer” is becoming increasingly refined. 1-3
WHAT IS CANCER?
In the normal eukaryotic organism, tissues achieve growth equilibrium, defined as the point at which production of new cells equals the death rate of cells, so that there is no net gain of tissue. In the case of tumors, cell replication exceeds physiologic need. Hyperplasia is the normal tissue’s response to certain noxious stimuli, but in contrast to neoplasia, hyperplasia is reversible upon return to the non-stimulated state. When expansion of cell numbers occurs in a locally confined form, the neoplasia is benign. Malignant neoplasia, or cancer, is unambiguously defined as the condition in which abnormal cells invade adjacent normal tissue or successfully colonize distant locations in the body through metastasis. 2
CANCER AS A GENETIC DISEASE
Cancer is considered a genetic disease because specific changes in the genome are necessary for expression of the malignant phenotype. Initiating changes that lead to malignant transformation typically involves regulation of the G1/S phase transition of the cell cycle. 4 When cell cycle regulation breaks down, increasing genetic errors and progression of the malignant phenotype is evident. The traditional understanding of carcinogenic genetic changes has focused on mutational events, such as point mutations and large rearrangements of chromosome material (translocation, gene amplification, and deletion). These alterations in the coding sequence may occur spontaneously, as seen in somatic gene recombination errors in lymphoma, or through errors in DNA polymerase nucleotide incorporation during DNA replication. In addition to these inherent stochastic risks for genetic alteration, cancer is also caused by the action of external carcinogenic agents such as chemical, physical or viral carcinogens, as will be discussed elsewhere in this book. 1-5 While changes in the coding sequences of DNA are essential to carcinogenesis, cancer also encompasses epigenetic alterations that do not involve mutating the DNA coding sequences. These epigenetic changes include gene silencing through DNA promoter methylation and histone acetylation, as well as through regulation of post-transcriptional and post-translational events 6 such as production of small interfering RNA molecules ( Table 1-1 ).
| Epigenetic Alterations | Cellular Response |
|---|---|
| DNA promoter methylation | Gene silencing 25 |
| Histone modification (methylation, acetylation) | Methylation → gene silencing 26 Acetylation → gene expression 27 |
| MicroRNAs | RNA expression modulation (oncogenic or tumor suppressor function) 28 |
Cancer cells and tissues are generally undifferentiated as compared with normal tissues of the same cellular origin, and the aggressiveness of biologic behavior correlates with the degree of anaplasia. In some senses, the study of cancer is very similar to developmental biology in reverse. In embryogenesis, cells are anaplastic or undifferentiated in the beginning, and subsequently differentiate as they move to the appropriate organ location or site of maturation. In cancer, a primordial stem cell, or a committed or partially committed cell with replicating potential, replicates inappropriately such that the fully mature cellular phenotype is not achieved. 7
WHICH CELLS ARE SUSCEPTIBLE TO MALIGNANT TRANSFORMATION?
Most researchers believe that cancers result from genetic derangement in a cell that retains replicative potential. The stem cell theory states that pluripotent stem cells, rather than terminally differentiated cells, are the most likely originators of cancer. In this schema, malignant cells generally arise from faulty differentiation of pluripotent or semi-committed cells that retain replicative potential, rather than from de-differentiation of a mature, terminally differentiated cell. However, de-differentiation of mature cells has also been documented to occur in certain malignancies. Cells that replicate more often during the lifetime of the individual are more prone to manifesting aberrant cell replication. Thus, cancers arise most commonly in cells that divide throughout the lifetime of the individual, and carcinomas (cancers arising from cells of ectodermal or endodermal origin) are numerically much more frequent than sarcomas (cancers arising from cells of mesodermal origin).
Some of the events leading to malignant transformation may be related to the environment in which a cell develops. For example, implantation of neural-crest cells from a developing mouse embryo into the testes of a normal mouse results in the development of teratocarcinomas. Conversely, incorporation of malignant teratocarcinoma cells into a developing mouse blastocyst results in the formation of a normal, healthy mouse that contains a mixture of genotypes derived from both the normal cells of the blastocyst and the teratocarcinoma. 8 These experiments demonstrate that epigenetic influences supplied by the tumor cell’s microenvironment are involved in cancer development, just as they are in normal cell differentiation in embryogenesis.
CLONALITY, HETEROGENEITY, AND CELL CYCLE REGULATION
Most tumors are clonal in origin; that is, tumors are derived from a single cell that has undergone malignant transformation. However, cells within the cancerous mass are also heterogeneous. Although they originate clonally, by the time a large tumor burden has developed, cancer cells have undergone further mutation and changes in karyotype. Also, cancers that arise from stem cells may have populations of cells representing various stages of differentiation as they attempt to follow more normal maturation pathways. Tumor cell heterogeneity renders treatment of tumors more difficult, since different cells have different growth rates and different sensitivities to chemotherapy and radiation therapy.
Cancers are progressive in nature and undergo what has been called a “bad to worse” phenomenon. This may be because one of the earliest genetic derangements to occur in cancer is the loss of cell cycle regulatory control. In the normal cell cycle, intracellular conditions must be optimum for replication to proceed. Cells that have DNA damage or lack of appropriate precursor nucleotides and proteins for cell replication will halt in progression through the cell cycle, typically at the G1/S phase transition checkpoint. Once DNA has been repaired and protein and nucleotide levels are determined to be sufficient, the cell resumes passage through the cell cycle. Progression through the cell cycle is determined by checkpoint regulators such as cyclins and cyclin-dependent kinases (CDKs), tumor suppressors such as the Rb gene, progression factors, DNA repair enzyme activity, cytoplasmic factors such as mitotic spindle assembly, and regulators of apoptosis (programmed cell death) ( Figure 1-1 ). 9 Defects in any of these systems can result in aberrant cell replication. One of the most common genetic defects detected in human cancers thus far is found in the p53 gene pathway. The p53 gene, along with its upstream and downstream signaling partners, is responsible for halting a deranged cell in the G1 phase of the cell cycle. If a normal cell is unable to complete repairs and prepare for replication within a short time, the cell will “commit suicide” (undergo apoptosis) for the benefit of the organism. Because defects occur so commonly in p53 and similar pathway regulation, cancer cells accrue many more mutations over time because they are unable to stop replicating to allow repairs to proceed ( Figure 1-2 ). 10 This phenomenon of early loss of the ability to correct for damaged DNA is referred to as the “mutator phenotype” of cancer cells.
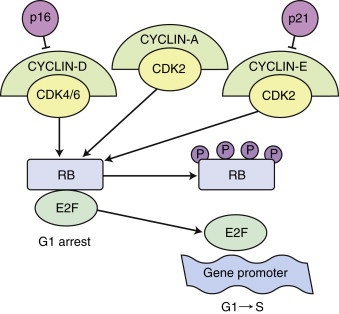 |
| FIGURE 1-1 The RB function in G1 to S phase during cell-cycle progression. Eukaryotic cells have evolved signaling pathways to coordinate cell-cycle transitions and ensure accurate replication of DNA before cell division. Cell-cycle progression is stimulated by protein kinase complexes, which consist of a cyclin and a CDK. The CDKs are expressed constitutively throughout the cell cycle, whereas cyclin levels are restricted by transcriptional regulation and by ubiquitin-mediated degradation. The activation of CDK requires the binding of a cyclin partner and a site-specific phosphorylation. The complexes cyclin-D/CDK4, cyclin-D/CDK6, cyclin-E/CDK2, and cyclin-A/CDK2 all contribute to the regulation of G1 to S phase progression. Unphosphorylated pRb is believed to bind to the transcription factor E2F and prevents E2F-mediated transcription of genes that are responsible for transition from the G1 to the S phase of the cell cycle. Phosphorylation of RB by cyclin-D/CDK4/6 and cyclin-A/-E/CDK2 complexes causes the release of RB from E2F, with subsequent G1 to S progression. Loss of pRb, synthesis of truncated pRb, or mutations that alter the ability of unphosphorylated pRb to bind to E2F, all allow unrestricted progression from G1 to S phase. |
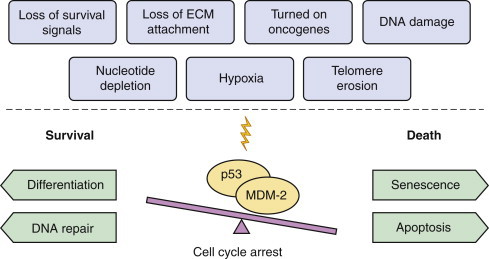 |
| FIGURE 1-2 The p53/MDM-2 balance. The p53 gene, along with its negative regulator MDM-2, mediate the cellular response to various stress signals. Whether the balance leans towards life (differentiation, DNA repair, cell cycle arrest) or death (senescence, apoptosis, cell cycle arrest) depends on several factors, including cell type, extracellular microenvironment, and other damages sustained in the cellular molecular machinery. The point of p53/MDM-2 balance is to favor the preservation of DNA integrity. |
CANCERS GO FROM BAD TO WORSE: THE MULTISTEP NATURE OF CARCINOGENESIS
Over time, cancer cells become more anaplastic, resistant to apoptosis, and more rapidly cycling. An example of this phenomenon is the genetic progression of colon cancer in humans. People who carry the genotype of familial colonic polyposis have a very high predisposition to develop colonic carcinoma. In an important study by Fearon and Vogelstein, 11 progressive genetic changes were mapped in the colon cells of individuals with a familial syndrome referred to as APC (adenomatous polyposis coli). Over time, colon cells of these individuals develop benign adenomatous polyps, pre-malignant carcinomas in situ , locally invasive but non-metastatic colon cancer, and finally aggressive metastatic disease. Sequential and predictable additive mutations are seen in these family members throughout the course of disease progression.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


