Fig. 6.1
Representative clips for donor (left) and recipient (right) preparation during the murine heart transplant surgery
6.
Make an incision on the posterior lateral thoracic cavity on the left and right sides of the chest.
7.
Place a loose 5-0 silk suture around the inferior vena cava (IVC).
8.
Insert it into the right atrium.
9.
Inject 1.0 cc of 4 °C heparinized saline into the IVC.
10.
Ligate the IVC with the 5-0 silk suture.
11.
Isolate the right superior vena cava (SVC) in a similar fashion.
12.
Ligate it with 5-0 silk suture.
13.
Roll the heart toward the animals’ right side and isolate the left SVC.
14.
Ligate and divide it with a 5-0 silk suture to expose the left pulmonary artery (PA).
15.
Use micro-scissors to divide the aorta proximal to the right brachiocephalic artery.
16.
Reflect the aortic cuff to expose the PA trunk and the left and right branches of the PA.
17.
Divide the PA trunk as distally as possible.
18.
Place a 5-0 silk suture around the base of the heart and tie.
19.
The heart is then cut free at the base and placed in 4 °C saline.
6.3.2 Heart Implantation into the Recipient Mice
1.
Anesthetize the recipient mice with pentobarbital.
2.
Immobilize the mouse using arm and leg restraints.
3.
Wipe the skin with alcohol and drape in a sterile fashion.
4.
Make a midline vertical abdominal incision and enter the abdominal cavity using clips (Fig. 6.1).
5.
Retract the bowel on to the chest and keep it wrapped in a sterile, moist gauze.
6.
Isolate the abdominal aorta and IVC below the renal vessels (Fig. 6.2).
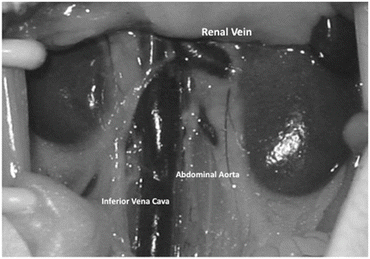
Fig. 6.2
A representative photo of the recipient abdominal aorta, inferior vena cava, and renal vessels
7.
Use 10-0 nylon suture to ligate any lumbar vessels within the field.
8.
Use micro-clips to nip the inferior followed by the superior vessels.
9.
Form an aortotomy with a 30 G needle in order to enter the lumen of the aorta.
10.
Extend the incision with fine micro-scissors to a length of approximately 2 mm.
11.
Perform an end-to-side anastomosis of the donor aorta to the recipient aorta in the following fashion.
12.
Place a 10-0 nylon suture stay stitch on the donor aorta and on the inferior angle of the incision in the recipient aorta and tie.
13.
Place a second 10-0 nylon suture opposite the first in the donor aorta and the superior corner of the incision in the abdominal aorta and tie.
14.
Make a running suture line from superior to inferior in the lateral wall of the aorta and tie with the previously placed stay stitch.
15.
Perform an end-to-side anastomosis of the donor pulmonary artery to the recipient IVC in the following fashion.
16.
Puncture the IVC with a 30 G needle and extend the incision for approx. 2 mm with fine micro-scissors.
17.
Tie the donor pulmonary artery to the inferior corner of the incision in the IVC with 10-0 nylon.
18.
Place a second 10-0 nylon opposite the first in the donor artery and the superior corner of the incision in the IVC and tie.
19.
Make a running suture line between the pulmonary artery and the IVC and tie.
20.
Release the distal micro-clip to reestablish venous flow.
21.
Once hemostasis of the venous anastomosis is observed, the proximal micro-clip is gradually loosened and the arterial anastomosis observed for hemostasis.
22.
When both anastomoses are considered secure, remove the micro-clips from the mouse (Fig. 6.3).
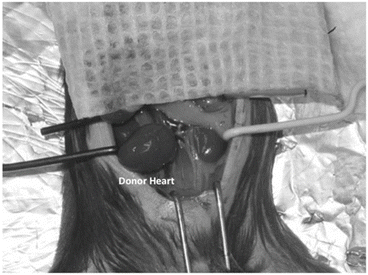
Fig. 6.3
A representative photo of the donor heart after transplantation
23.
Return the bowel to the abdomen.
24.
Use 6-0 silk suture to close the abdominal wall in two layers.
25.
Recover the animal on a warming blanket.
6.4 Pathology of GAD
6.4.1 Immunological Features and Risk Factors
Pathologically, GAD involves the entire length of the transplanted arterial vasculature. GAD is characterized by concentric intimal thickening comprised of SMCs and ECM, with relatively intact internal and external elastic laminae. The vascular media and adventitia are relatively unaffected, while inflammatory cells are variably present in evolving lesions. Calcification and atheroma formation are also uncommon. It is known that the clinical risk factors for GAD are as follows: donor history of hypertension, induction therapy with IL-2 antagonist or OKT3, pretransplant diagnosis of coronary artery disease, increasing number of HLA-DR mismatches, and so on. Other risk factors include obesity, diabetes, hyperlipidemia, viral infection, and hyperhomocysteinemia [3].
Allografts are subject to multiple injuries, such as immune and nonimmune factors. Although many pathological factors contribute to GAD development, any of the various injuries can independently induce GAD. Immune injury is known to play a critical role in GAD pathogenesis. Although cellular immune responses have a central role in GAD development, humoral injury is also recognized as a critical component. The predominant effector cells are macrophages; they secrete proinflammatory cytokines that influence smooth muscle-like cell (SMLC) and fibroblast proliferation. It also enhances ECM synthesis which results in GAD formation. Recipient T cells require two signals to be activated; one signal involves host T-cell receptor interaction with the donor major histocompatibility complex, and another signal involves ligation of costimulatory molecules. The nature and efficiency of both signals affect the development of GAD [14, 15]. Nonimmune risk factors, including ischemia, diabetes, and hypertension, correlate with intimal thickening of GAD. Periods of ischemia are significantly important for promoting GAD; these effects are attributable to ischemic EC, which increases local platelet accumulation. EC ischemic injury can also increase the major histocompatibility complex and adhesion molecule expression [16, 17]. Cytomegalovirus is known to be associated with an increased incidence of GAD, which is linked to wound repair and angiogenesis [18, 19].
6.4.2 Altered Myosin Heavy Chain Isoforms
We reported that there are three types of smooth muscle myosin heavy chain (MHC) isoforms expressed in vascular SMCs: SMl, SM2, and SMemb. SMl is expressed throughout early developmental stages to mature stages, and SM2 is expressed only after birth. These two isoforms are specific to SMCs, whereas SMemb is a non-muscle-type MHC isoform that is expressed in embryonic SMCs of fetal aorta and proliferating SMCs. The expression of these MHC isoforms can be a useful molecular marker for detection of pathological conditions in acute or chronic rejection. We showed that the coronary arteries had significant intimal thickening, as indicated in pathological samples in rat allografts taken on day 35 from FK506-treated recipients. Monkey allografts harvested on day 44 treated with anti-leukocyte function-associated antigen (LFA)-1 mAb showed severe GAD with perivascular cell infiltration. Immunohistochemically, SM2 was positive in the media of coronary arteries in native rat hearts and isografts. In FK506-treated rat allografts harvested on day 35, SM2 expression was eliminated in both the media and thickened intima. However, a reduced SM2 expression was also observed in the media, even in the area lacking the intimal thickening in the FK506-treated rat allografts on day 14. The expression of α-SMA was observed both in the media and in the proliferative intima of all the coronary arteries (Fig. 6.4). In native monkey hearts, SMemb was negative in the media of coronary arteries. However, SMemb was expressed in thickened intima and media in monkey coronary arteries harvested on day 44 (Fig. 6.5). Transmission electron microscopy showed severe intimal thickening of graft coronary arteries in the allograft from FK506-treated rats on day 35. Thickened intima consisted of phenotypically modulated SMCs: some had abundant actin filaments, caveolas, and dense bodies similar to the normal phenotype. Others showed abundant mitochondria and endoplasmic reticula. Reduced actin filaments demonstrated phenotypic modulation (Fig. 6.6). We demonstrated that chronic rejection is characterized by diffuse coronary arteriosclerosis formed by diffuse intimal thickening that consists of proliferative vascular SMCs with phenotypic modulation. Because the proliferation of SMCs is regulated by sustained immune responses, the phenotypic modulation of SMCs can be a sensitive indicator of chronic rejection. In the study, we demonstrated that enhanced SMemb expression and reduced SM2 expression in the media as well as in the thickened intima of coronary arteries were shown in chronically rejected cardiac allografts. These cells were phenotypically modulated SMCs with proliferative states characterized by abundant organelles on electron microscopy, indicating the altered expression of MHC in early chronic rejection. Altered MHC expression was also observed in the media even in the area without intimal thickening. The usefulness of this approach in sensitive detection of phenotypic modulation, before morphological changes occur, was clearly shown. We also noticed altered MHC expression present simultaneously in both endomyocardial arterioles and epicardial coronary arteries of chronically rejected allografts. This result suggested that chronic rejection could be demonstrated by altered MHC isoform expression in endomyocardial arterioles. Therefore, altered MHC expression in GAD is an early sign of chronic rejection [10].
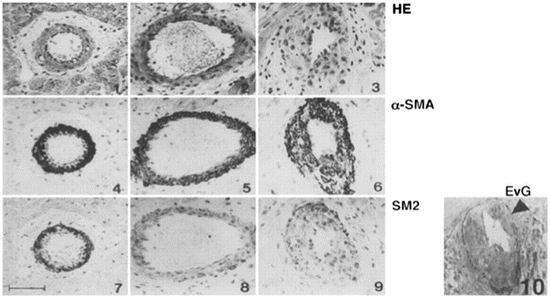
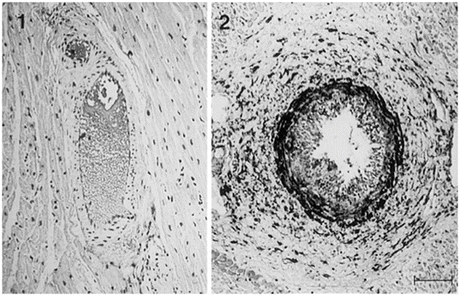
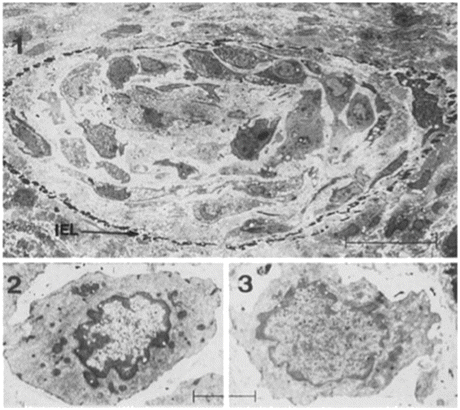
Fig. 6.4
Microscopic findings of rat coronary arteries. Panels at top (1 through 3) were stained with hematoxylin and eosin (HE), those in the middle (4 through 6) with anti-α-SMA antibody, and those at bottom (7 through 9) with anti-SM2 antibody. Elastica van Gieson staining was used to demonstrate intimal thickening (10). Both α-SMA and SM2 are expressed in the cells of the medial layer of native heart (left, 1, 4, and 7). The allograft harvested on day 14 with FK506 treatment (middle, 2, 5, and 8) does not show intimal thickening; however, SM2 expression in the cells of the medial layer was reduced. The allograft harvested on day 35 with FK506 treatment (right, 3, 6, 9, and 10) shows intimal thickening, and SM2 expression was also reduced in the medial layer and thickened intima. An arrowhead in panel 10 shows internal elastic lamina. Scale bar = 50 μm (From Ref. [10])
Fig. 6.5
Microscopic findings of monkey coronary arteries stained with anti-SMemb antibody (1, native monkey heart; 2, transplanted monkey heart with anti-LFA-1 mAb treatment harvested on day 44). Although SMemb was not expressed in the native monkey heart, SMemb-positive cells were observed not only in perivascular lesions but also in the thickened intima and media in the transplanted monkey heart. Scale bar = 50 μm (From Ref. [10])
Fig. 6.6
Transmission electron microscopic findings of a coronary artery occluded by intimal thickening (1, transplanted rat heart with FK506 treatment on day 35) (IEL indicates internal elastic lamina). The smooth muscle cells that composed the thickened intima demonstrated phenotypes similar to normal cells (2) or phenotypic modulation (3). 1, scale bar = 10 μm; 2 and 3, scale bar = 1 μm (From Ref. [10])
6.4.3 Costimulatory Signals
Costimulatory signals are important for stimulation of T cells and inflammatory cell recruitment into allografts [20]. It has been reported that GAD correlates with expression of adhesion molecules, including intracellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, E-selectin, and P-selectin [21, 22]. Besides facilitating EC interactions, lymphocyte integrins can also act as costimulatory molecules during antigen presentation. Although blockade of ICAM-1 and LFA-1 interactions induced allograft tolerance [23], the effect of the signal blockade on GAD development was not clarified. To prove the effect, we treated C3H/He mice that received cardiac allografts from BALB/c mice with anti-ICAM-1 plus anti-LFA-1 mAbs for the first 5 days after transplantation. For control studies, FK506 was administered daily to other allograft recipients. Allografts were harvested on day 60.Immunohistochemical analysis was used to detect the expression of ICAM-1 and VCAM-1, and in situ reverse transcriptase–polymerase chain reaction (RT-PCR) was performed to detect platelet-derived growth factor (PDGF)-B mRNA expression in the graft arteries. We found that allografts from mice that received FK506 treatment daily showed significant neointimal thickening with increased expression of ICAM-1, VCAM-1, and PDGF-B mRNA, whereas there was almost no intimal thickening and ICAM-1, VCAM-1, and PDGF-B mRNA expression in the mice that received anti-ICAM-1 plus anti-LFA-1 mAbs. We concluded that short-term blockade of ICAM-1 and LFA-1 adhesion not only induced immunologic tolerance to cardiac allografts but also prevented GAD [24] (Fig. 6.7).
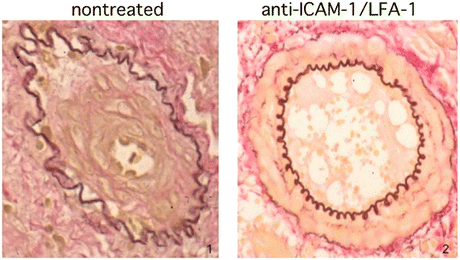
Fig. 6.7
Representative GAD findings of nontreated (1) and anti-ICAM-1 and LFA-1 antibody treated (2) arteries are demonstrated (From Ref. [24])
It has also been reported that cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) blockade of B7-CD28 interactions attenuated GAD [25]. The inducible costimulator (ICOS) molecule on T cells could also influence GAD development by directly interacting with ICOS-ligand on medial SMC activated by interleukin (IL)-1β [26, 27]. We also revealed that blockade of selectin [28], LIGHT [29], and 4-1BB [30] attenuated GAD, whereas programmed death-1 (PD-1) blockade accelerated the development [31]. Because each costimulatory signal has a different effect on GAD development, the anti-costimulatory therapy should investigate different organs and conditions.
6.4.4 Cytokines and Chemokines
Cytokines mediate local and systemic immune responses. Interferon (IFN)-γ is the most important player because it activates macrophages, augments inflammatory cell recruitment, and amplifies immune responses [32]. Despite ongoing acute rejection, the absence of IFN-γ abrogates GAD [33]. In association with the IFN-γ-induced cytokines, the IFN-γ and IL-12 relationship is a critical element in GAD pathogenesis, because medial SMCs synthesize IFN-γ following stimulation by IL-12 and IL-18 [34]. Although IFN-γ blocks medial SMC proliferation in vitro, IFN-γ directly induces intimal hyperplasia in the absence of inflammatory cells [35]. Other proinflammatory cytokines downstream of IFN-γ also play an important role.
Among the proinflammatory cytokines, tumor necrosis factor (TNF) also plays a pivotal role. It is well known that TNF promotes multiple aspects of allograft rejection via binding to type 1 (p55) and type 2 (p75) receptors. To reveal the role of TNF and its receptors on GAD formation, we used TNF type 1 receptor-deficient (TNFR1KO), type 2 receptor-deficient (TNFR2KO), and receptor double-deficient (TNFRDKO) mice to assess the relative roles of TNFR in acute rejection and GAD. Heterotopic cardiac transplantation was performed between C57BL/6 and Balb/c mice (total allomismatch) to assess the effects on graft survival; C57BL/6 and B6.C-H2<bm12>KhEg mice (Bm12, class II mismatches) were used to assess the effects on GAD 8 weeks after transplantation. We found that graft survival in the total allomismatch combinations was the same regardless of TNF-receptor status. In class II mismatches, wild-type (WT) combinations showed severe GAD, and GAD was not diminished when WT hearts were transplanted into TNFRDKO hosts. TNFR1KO donors or TNFR2KO donors had GAD comparable to WT donors; however, GAD was significantly diminished in B/6 TNFRDKO donor hearts. Because host TNF-receptor deficiency did not reduce GAD, the effect was not attributable to TNF signaling on host inflammatory cells. We conclude that both p55 and p75 signals on donor vascular wall cells are involved in the development of GAD, and either TNF receptor is capable of mediating a response that will culminate in GAD [36] (Fig. 6.8).
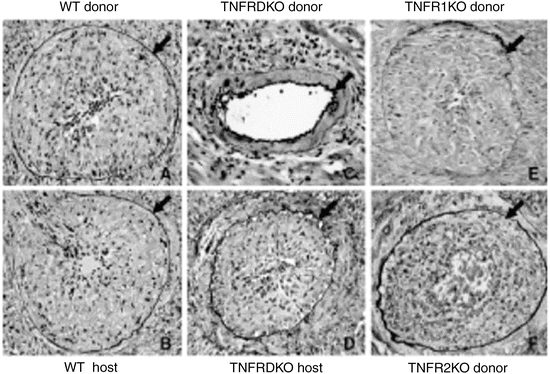
Fig. 6.8
Representative GAD findings are shown. Severe GAD develops by 8 weeks in both WT B/6 donor hearts in Bm12 hosts (a), as well as in Bm12 hearts transplanted into WT B/6 hosts (b). Major histocompatibility complex class II-mismatched TNFRDKO B/6 allografts implanted into WT Bm12 recipients showed significantly less GAD (c) compared to WT B/6 donor hearts. However, the WT Bm12 allografts implanted into TNFRDKO recipients showed severe intimal thickening (d), comparable to WT B/6 hosts. Both TNFR1KO (E) and TNFR2KO (F) donor hearts transplanted into Bm12 hosts showed severe GAD comparable to WT combinations (arrows indicate internal elastic lamina; original magnification, 200X) (From Ref. [36])
Chemokines are known to be recruiting and activating inflammatory cells. They are classified in four groups, CC, CXC, XC, and CX3C. The roles of these molecules in acute rejection and GAD have been investigated [37]. RANTES is a well-characterized CC chemokine implicated in both atherosclerotic and GAD pathogenesis [38]. Further chemokines, the IFN-γ-inducible IP10, Mig, and I-TAC, are critical for their strong association with GAD [39, 40]. Moreover, targeted deletion of CCR1 reduced both acute rejection and the development of GAD [41].
6.4.5 Sources of Cells in GAD
Although SMLCs are similar in appearance to medial SMCs, they are phenotypically modulated cells with different genetic expression profiles. We revealed that medial SMC exhibits a contractile function, whereas SMLC exhibits a more embryonic, synthetic phenotype [10]. Previously, GAD intimal cells were assumed to derive from the migration and proliferation of donor medial SMC. However, several groups have shown that intimal SMLC can also be of host origin. In animal models, the proportion of host origin SMLC in intimal lesions exceeds 90 % [42–44]. However, in gender-mismatched cardiac allografts, Y-chromosome in situ hybridization identified only 3–15 % host-derived intimal SMLC [45, 46]. These results demonstrate that both host bone marrow and non-bone marrow precursors contribute to the GAD development. Therefore, therapies against GAD may need to account for different cells of origin, routes of recruitment, and differentiation pathways.
6.5 Possible Agents for GAD Prevention
6.5.1 Immunosuppressants
Cyclosporin A (CyA) significantly suppressed acute cardiac allograft rejection; however, it was not effective in preventing GAD [47]. Although tacrolimus provides superior prevention of acute rejection compared with CyA, GAD development was similar between CyA- and tacrolimus-treated groups [48]. It has been reported that mycophenolate mofetil (MMF) significantly decreased first-year maximal intimal thickness [49] and the incidence of GAD [50]. Recently, proliferation signal inhibitors are known to have significant effects for preventing GAD. Sirolimus delays the progression of GAD in some clinical trials [51, 52]. Another proliferation signal inhibitor, everolimus, also prevented GAD development [53]. However, as there is no report to demonstrate the long-term effect of proliferation signal inhibitors to suppress GAD, further observation studies are needed.
6.5.2 AntiHypertensive Drugs
Calcium channel blockers are known to delay the progression of GAD and enhance coronary vasodilation [54]. Angiotensin-converting enzyme (ACE) inhibitors partially improve allograft endothelial dysfunction, oxidative stress, and endothelin activation. They are also associated with plaque regression and improved graft survival [55, 56]. Recently, a synergistic beneficial effect of a calcium channel blocker and an ACE inhibitor on the development of GAD has been described [57].
We showed that an effect of telmisartan, a potent angiotensin II receptor blocker, was attenuating GAD in murine cardiac allografts. Recently, it was reported that telmisartan could function as a partial agonist of peroxisome proliferator-activated receptor-γ (PPAR-γ) in addition to a blocker of angiotensin II receptor. We investigated the effect of telmisartan on chronic rejection. Hearts from Bm12 mice were transplanted into C57BL/6 mice (class II mismatch), and allografts were harvested at 8 weeks after transplantation. Recipient mice were fed either control chow or chow containing telmisartan from 1 day before transplantation. Proliferation assays of SMCs, which were isolated from the aorta of B/6 mice, were performed. Although severe neointimal hyperplasia developed in allografts from control mice fed chow, neointimal hyperplasia was significantly attenuated in allografts from mice fed chow containing telmisartan. Expression of IFN-γ and IL-15 mRNAs and MMP-2 in allografts was significantly lower in telmisartan-treated mice than in control mice. Proliferation of SMCs in response to fetal bovine serum was suppressed significantly by telmisartan. The PPAR-γ antagonist blocked telmisartan-induced suppression of SMC proliferation. Thus, telmisartan attenuates SMC proliferation via PPAR-γ activity and suppresses neointimal hyperplasia after transplantation. Telmisartan may be useful for suppressing chronic allograft rejection [58].
6.5.3 HMG-CoA Reductase Inhibitors
Studies have shown that HMG-CoA reductase inhibitors (statins) play an important role in the prevention of GAD. The effects of statins are mediated by multiple immunogenic effects. Simvastatin reduced proinflammatory cytokine activity, improved coronary endothelial function, and increased coronary lumen area [59]. We demonstrated that pitavastatin has an effect for prevention of GAD in an animal model. We investigated the role of pitavastatin, a potent HMG-CoA reductase inhibitor, in cardiac allograft rejection and mechanism of GAD suppression. Balb/c mice hearts were transplanted into C3H/He mice (a full allomismatch combination) to assess acute rejection or C57BL/6 hearts into Bm12 (a class II mismatch combination) to examine the extent of GAD. Pitavastatin was orally administered to mice everyday. To assess the effect in acute rejection, mixed lymphocyte reaction (MLR) was performed and cytokine mRNA expression was examined by ribonuclease protection assay (RPA). We found that pitavastatin significantly prolonged allograft survival. Lymphocyte proliferation was inhibited by pitavastatin, and RPA showed downregulation of IL-6 in pitavastatin-treated cardiac allografts. Allografts in the pitavastatin-treated group after 8 weeks showed less GAD compared to the control group. In an in vitro study, pitavastatin suppressed the SMC proliferation in response to activated T cells and inhibited extracellular signal-regulated kinase 1/2 activation. Thus, we concluded that pitavastatin could be effective to suppress GAD development in cardiac transplantation [60] (Fig. 6.9).




