Mouse models
Diets
Reported duration for atherosclerotic lesion formation
Reference no.
Apoe−/−
Normal chow diet
16–20 weeks old
[72]
Human ApoE*3 Leiden transgenic
High-fat/high-cholesterol diet
16 weeks
[20]
Ldlr−/−
High-fat/high-cholesterol diet
16–20 weeks
[13]
Ldlr−/− x human apoB-100 transgenic
High-fat/high-cholesterol diet
24 weeks
[73]
Ldlr−/− x Apobec-1−/−
Normal chow diet
8 months (32 weeks) old
[17]
Apoe−/− x Ldlr−/−
Normal chow diet
14 weeks old
[18]
Mouse Apoa2 transgenic
High-fat/high-cholesterol diet
11–12 weeks
[74]
Human apoB transgenic
High-fat/high-cholesterol diet
18 weeks
[75]
Ldlr−/− x human CETP transgenic
High-fat/high-cholesterol diet
3 months (12 weeks)
[22]
Apoe−/− x human CETP transgenic
High-fat/high-cholesterol diet
3 months (12 weeks)
[76]
Human apoE*3 Leiden transgenic x human CETP transgenic
High-fat/high-cholesterol diet
19 weeks
[77]
Human apoB transgenic x human CETP transgenic
High-fat/high-cholesterol diet
20 weeks
[23]
PCSK9 gain-of-function mutants (adeno-associated viral vectors)
High-fat/high-cholesterol diet
12 weeks
[26]
SR-BI KO/ApoeR61(h/h)
High-fat/high-cholesterol diet
4 weeks
8.3 Atherogenic Diets
Depending on the main purpose of each study, different strategies to induce atherosclerosis in mice may be used. A series of diets are commercially available (Research Diets, Harlan Laboratories, and Purina Diets), which promote the development of atherosclerosis. Here, we review the common types that have been used in many studies. It should also be noted that an appropriate choice of a mouse model is important in conjunction with the diet for successful study.
Normal chow diet, or otherwise known as the “control diet” in animal experiments, provides sufficient nutrition. Usually composed of ground ingredients such as corn, oats, wheat, and soybean meal, this grain- or cereal-based diet is designed to meet the requirements of a healthy rodent diet (along with other additives such as vitamins, minerals, and fat). An animal group fed with an atherogenic diet may be paired with another group with a control diet with no added cholesterol and fat. Such a control diet should ideally use purified chow rather than regular chow, as contents of regular chow vary depending on the season, which may influence on the results of experiments [30].
The Western diet is a purified diet composed of 20 % milk fat/butter fat, 34 % sucrose, and 0.2 % cholesterol and is widely available. Fat and cholesterol amounts can be modified to higher levels. It is popularly used with genetically manipulated mouse strains, such as Ldlr−/− and Apoe−/− and others.
Another common diet is the “Paigen” atherogenic diet (a hybrid diet created by mixing a natural ingredient mouse diet in a 3:1 ratio with “Thomas-Hartroft” diet) which consists of 16 % fat (half derived from cocoa butter), 1.25 % cholesterol, and 0.5 % sodium cholate.
“Clinton/Cybulsky” AIN-76A semi-purified high-fat diet which contains 17 % cocoa butter and 1.25 % cholesterol (category: D12108C, Research Diets) compared to the “standard” AIN-76A rodent diet.
To effectively induce quantifiable atherosclerotic plaques in mice, it is crucial to understand and pick which nutrients you may wish to add/deduct from the rodent diet before starting the administration. Addition of dietary cholesterol is a key factor for promoting atherosclerosis in especially some genetically modified mouse strain [31]. Ishibashi et al. have indicated that the greater the increase in lipid-rich diet in Ldlr−/− mice, the greater the increase in plasma cholesterol can be achieved [32]. Added dietary cholate for atherosclerosis studies in LDLR −/− mice was reported to be controversial due to its toxic hepatic metabolic effects [33].
8.4 Feeding Study Protocol
Before starting any animal experiment, approval of the experiment via animal research protocol by a local institutional committee for animal care and use/NIH is required. All federal regulations and institutional policies need to be fully abided.
The materials needed for an administration protocol are genetically altered mice (e.g., Ldlr−/−, Apoe−/−) and wild-type mice (for control group) of at 6–10 weeks of age (and at least 18 g in body weight) and the prepared diet. Depending on goals of each study, the diet can be composed of either chow or high-fat diet with/without additive cholesterol. The diet can be customized as well to test a compound of interest or a drug. For safer measures, it is always recommended to have the diet irradiated for sterility before administering it to the animals. The prepared diets should last for 3–6 months in cold storage, but if testing out a drug/compound, one needs to take into account the shelf life of the compound mixed in with the diet. The activity level of the compound may also alter when going through the milling process (heating and cooling).
With 6–10-week-old Ldlr−/− mice, high-fat/high-cholesterol diet administration (ad lib) should be between a minimum of 4 weeks and ~20 weeks (study end point) to induce substantial levels of plaque formation within the aorta and carotid arteries. Apoe−/− mice on the other hand do not require such extensively long diet administration to see similar amount of plaque formation, due to the strain’s trait of possessing severe spontaneous hypercholesterolemia earlier in age, in comparison to its Ldlr−/− counterpart. Even if fed with a milder “high-fat” diet, Apoe−/− mice are known to induce severe hypercholesterolemia [34].
8.5 Ultrasound Imaging of the Carotid Artery
The development of high-resolution ultrasound imaging has helped translational and preclinical researchers noninvasively visualize in vivo tissue structure and physiology [35]). This makes it an excellent and clinically relevant tool for small animal imaging to measure surrogate end points. High frame rate ultrasonography and advanced echocardiography have been made accessible to laboratories worldwide largely due to the efforts of VisualSonics through their line of high-resolution micro-ultrasound machines (e.g., Vevo 2100, Vevo LAZR, Vevo 3100). Imaging the vasculature, including the aorta, brachiocephalic, and carotid arteries, with intact physiologic blood flow, is critical for accurate morphometric analyses in mouse models of atherosclerosis. High temporal resolution accorded by this modality provides data such as vascular wall motion velocities, displacement, distensibility, and stiffness through a speckle-based tracking algorithm in VisualSonics’ Vevo®Vasc Advanced Analysis software package [36]. Their high-frequency transducers (e.g., MS 700 of the Vevo 2100) combined with a stepping motor (minimum of 10 μm steps) can produce detailed 3D image data set from serial slices of 2D B mode, color Doppler, contrast, or power Doppler images. Targets such as the neointima can be segmentally traced for accurate in vivo volumetric measurements. In addition, the MS 700 having a center frequency of 50 MHz and bandwidth of 30–70 MHz can image the mouse carotid artery with an axial resolution of 30 μm and a lateral resolution of 70 μm. A clear delineation of the intima-media border can be visualized in the atherosclerotic vessel, which appears as a thin black line “sandwiched” between a slightly hyper-echoic (gray) adventitia and a very hyper-echoic (white) endothelial/intima layer.
Following protocol is an example of acquiring a short-axis and a long-axis B mode, ECG-based kilohertz visualization (EKV) image, and 3D B-mode image of the mouse carotid artery.
Materials
Vevo 2100 and MS 700 Transducer (VisualSonics)
Vevo Integrated Rail System III (VisualSonics)
ECG platform and physiologic monitor
Transducer clamp
Oxygen/medical air tank
Inhalational anesthesia system
Isoflurane
Transmission gel
Lubricant
ECG electrode cream
Depilatory cream
Cotton-tipped applicators
Kim wipes
Mice on pro-atherogenic or the control diet
Protocol
1.
Turn on the Vevo 2100 machine and allow it to fully initiate.
2.
Attach physiologic monitor to the Vevo 2100 and turn it on. Attach MS 700 to the transducer port and then initialize the scanhead.
3.
Set the platform temperature to 37 °C on the physiologic monitor.
4.
Fill to 80 % capacity the anesthesia vaporizer chamber with isoflurane and open the air tank.
5.
Set air flow to 1 l/min and isoflurane output to 2–3 %
6.
Place the mouse inside the anesthesia induction chamber. Secure airflow lines and direct isoflurane gas to the chamber. Allow 3–6 min for full sedation.
7.
Transfer the mouse onto the ECG platform in the supine position. Fit the animal’s snout into the nasal cone. Switch the isoflurane line toward the cone and reduce isoflurane to 1–1.2 %.
8.
Place ECG electrode cream onto the four metal ECG leads on the platform and then secure the paws of the mouse onto these leads using tape.
9.
Coat the rectal temperature sensor with lubricant before gently inserting into the mouse to monitor its temperature.
10.
Check the physiologic monitor and confirm the heart rate remains between 400 and 600 beats per minute and the temperature is close to 37 °C. For heart rate <400 bpm, reduce isoflurane to 0.8–0.9 %.
11.
Make sure the animal’s respiratory rate (RR) is between 50 and 80 cycles per minute. If <40 cpm, reduce the isoflurane. RR and ECG tracing can easily be seen on-screen during live scanning.
12.
Apply depilatory cream onto the neck region of the mouse from the submandibular area down to the sternal area extending to the lateral edges of the mouse’s neck. Massage the cream onto the fur using the cotton-tipped applicator and wait for 3–6 min.
13.
Remove the cream with detached fur using wet Kim wipes. Continue wiping with fresh wipes until all cream is gone revealing a hairless neck area. Dry the area.
14.
Place transmission gel onto the neck area and position the scanhead where the midportion of the common carotid artery is located, 0.5–1.0 cm above the skin of the neck. Make sure the horizontal plane of the transducer is perpendicular to the axis of the carotid.
15.
Press scan button to image live. The image is short-axis view or transverse section of the artery.
16.
The notched ridge on one side of the scanhead orients the imager to the image seen. This ridge corresponds to the vertical line beside a small dot located in the top left corner of the image window. This orients the probe position relative to the image.
17.
Locate midsection of the common carotid artery, which appears as a circular object pulsating on each side of the midline trachea. Change to color Doppler to confirm arterial pulsations inside the arterial lumen. If no pulsations are seen, adjust the pulse repetition frequency (PRF) to a lesser value using the “velocity” turn-knob.
18.
Center the target in the image window and reduce image width and depth while pushing the transducer nearer to the artery so as to “zoom in” on the artery. Adjust image focus using focal zone-type and depth rocker switches.
19.
Maximize the differential appearance (echogenicity) of the neointima, media, and adventitia of the artery by reducing the transmission power to the desired level while keeping the gain at 32 dB.
20.
Record cine loop of several frames (e.g., 300 or 800 frames). To get an EKV image, make sure the heart rate is regular and then switch to EKV acquisition mode. The EKV image is much more polished than a B-mode image. Its clarity depends on the EKV acquisition frequency which can range from 1000 Hz to 10,000 Hz. A higher EKV frequency will have longer acquisition time. The resulting EKV image is a reconstruction of multiple successive M-mode lines of image taken from the left edge of the acquisition window all through the right edge for every time frame within the ECG cycle.
21.
One can also take an M-mode image of the cross section. This allows acquisition of very high temporal images of the anterior and posterior vessel wall as it moves during several cardiac cycles. This movement and displacement may be affected by arterial distensibility, elasticity, or stiffness. Simply place the M-mode sector sampler exactly at the middle of the arterial section. Record cine loop.
22.
For a long-axis B-mode image, the probe is simply rotated in place by 90°. During B mode, the image window is adjusted by increasing the image width until the carotid bifurcation is seen. The artery should be in the most horizontal position for imaging. Any tissue plane is most clearly seen when they are directly perpendicular to the axis of insonation (long axis of the transducer). Adjust the mouse platform accordingly.
23.
Record cine loop and an EKV image if desired.
24.
All B-mode and EKV images can also be processed offline using the Vevo®Vasc Advance Analysis software for speckle tracking to measure several parameters like intima-media thickness (IMT); distensibility; elastance; shear stress; pulse propagation velocity; sector- or layer-specific motion parameters like radial, rotational, and tangential velocity; displacement; and lumen diameter changes.
25.
To get a 3D image of the artery, attach the Vevo 3D stepping motor to the rail system with the transducer clamp attached below the mini rail of the stepping motor. Position the transducer accordingly. Attach the motor connector cable to the back panel of the Vevo 2100 and initiate the motor through the software.
26.
Locate the artery and center it as closely as possible.
27.
Press the 3D button.
28.
Set up the 3D mode using the guide below:
3D parameter | Description |
|---|---|
Scan distance | Distance in millimeters that the 3D motor stage will traverse for image acquisition |
Step size | Sets the incremental distance between successive 2D images which affects the amount of detail contained in the 3D image and acquisition time |
29.
Start scanning. Once done, store cine loop.
30.
Load the 3D image as directed in the screen and go to 3D settings for volume rendering. Start tracing of the neointima or the lumen, proceed through the slices, and end as prompted on the screen. Volume will be automatically calculated. Various options in the screen can be used to show different 3D renderings and viewpoints.
8.6 Molecular Imaging
Molecular or functional imaging is a promising tool to visualize biological processes at molecular levels. Technical innovations, such as nanotechnology, have enormously contributed to the development of molecular imaging [37]. Macrophages play central roles in the pathogenesis of atherosclerosis from the initiation to the development and to the onset of acute thrombotic complications of atherosclerosis. Therefore, imaging of macrophages may help to further understand the mechanisms for atherosclerosis. Macrophage imaging may also serve as a powerful preclinical tool to monitor the effects of new therapies. This section briefly reviews several key modalities for molecular imaging of macrophages—magnetic resonance imaging (MRI), nuclear imaging, and near-infrared fluorescence (NIRF) imaging—and then describes a specific protocol of NIRF imaging in macrophage activities in atherosclerotic plaques.
8.6.1 Macrophage Imaging by MRI
Magnetic nanomaterials with iron oxide cores are used as contrast agents for MRI [37]. Superparamagnetic iron oxide (SPIO) nanoparticles and ultrasmall SPIO (USPIO) nanoparticles (<50 nm) represent such materials. Morishige et al. used 3.0 Tesla MRI and USPIO to report that, in rabbit atherosclerosis model, (1) changes of MRI signal intensity correlate with histologically evaluated macrophage accumulation and (2) macrophage imaging with USPIO can monitor the effects of lipid-lowering therapy on plaque [38].
8.6.2 Macrophage Imaging by Nuclear Medicine
Nuclear medicine is a promising modality to visualize macrophage burden and activity in atherosclerotic plaques in patients. PET with 18F-fluorodeoxyglucose (FDG-PET) can visualize cells with active glucose metabolism—macrophages in atherosclerotic plaques—and monitor the effects of drugs [39–41]. FDG-PET is also used in the field of preclinical research. FDG-PET can detect foamy macrophages in atherosclerotic plaques in hyperlipidemic rabbits [42]. Furthermore, a recent study reported new radionuclides targeting mannose receptor, a macrophage product [43]. Macrophages took up 2-deoxy-2-[18F]fluoro-D-mannose (18F-FDM) more efficiently than 18F-FDG, suggesting that 18F-FDM serves as a novel macrophage-targeting tracer.
8.6.3 NIRF Imaging of Macrophages
NIRF with a wavelength of 600–900 nm has several advantages for noninvasive imaging. The absorbance of NIRF signals by H2O or hemoglobin, which abundantly exists in human bodies, is small and thus penetrates the tissue. By combination with other modalities such as an angioscope, information about tissue surface appearance and protease activity in deeper tissues could be obtained simultaneously [44, 45]. The Jaffer and Tearney groups have developed an intravascular molecular imaging modality [46, 47]. This technology involves a catheter-assisted dual-imaging approach—optical frequency domain imaging (OFDI) and NIRF imaging. This dual-modality intra-arterial catheter can visualize atherosclerotic plaques with high protease activity in the rabbit aorta and porcine coronary arteries and has been in clinical development. In mouse models of atherosclerosis, NIRF images could be acquired by intravital confocal laser microscopy or fluorescence reflectance imaging (FRI) with appropriate laser and detector for NIRF.
8.6.3.1 Macrophage Accumulation
NIRF probes for macrophages are commercially available. AminoSPARK (PerkinElmer) consists of nanoparticles with iron oxide core and is coated with biocompatible polymer. AminoSPARK is taken up by macrophages, thus enabling to visualize phagocytic activity in vivo.
8.6.3.2 Proteinase Activity
Plaque macrophages produce various proteinases including matrix metalloproteinases (MMPs) and cathepsins. The evidence suggests that such matrix-degrading enzymes reduce collagen content in atherosclerotic arteries and weaken mechanical strength of the atherosclerotic plaques [48, 49]. Experimental studies used genetically altered mouse strains to support this theory [50, 51]. Preclinical studies including our own established the feasibility of molecular imaging of matrix-degrading enzymes in atherosclerosis [37, 52, 53].
8.6.3.3 Calcification
Human arteries and aortic valves often contain calcification. Although conventional views considered cardiovascular calcification as an aging-associated passive degenerative process, the recent evidence has recognized that calcification involves active biological processes closely linked with inflammation [16, 54, 55]. In humans, the recent evidence has indicated that coronary artery calcium (CAC) score detected by CT predicts future cardiovascular events [44, 56]. Preclinical evidence suggests that early calcification (“microcalcification”) reduces atherosclerotic plaque stability and increases the risk of disruption and thrombus formation [57]. Molecular imaging with the NIRF probes OsteoSense 680 for osteogenic activity and AminoSPARK for macrophages, co-injected in atherosclerotic mice, demonstrated that macrophage accumulation precedes calcification (Fig. 8.1a) [54]. The same study also demonstrated that statin treatment retarded the development of macrophage burden and calcification in parallel (Fig. 8.1b), indicating that anti-inflammatory therapies may prevent calcification and thus cardiovascular events. Figueiredo JL et al. demonstrated that cathepsin S inhibition decreases osteogenic activity in atherosclerotic lesion of Apoe−/− mice [58].
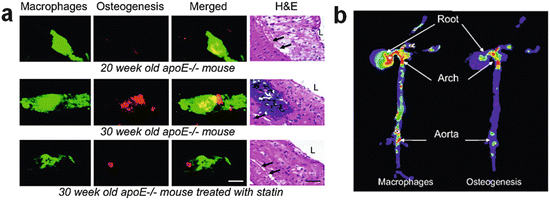
Fig. 8.1
NIRF imaging of macrophages and calcification in mouse atherosclerotic plaques. (a) Intravital NIRF imaging of the left carotid artery of Apoe−/− mice for osteogenic activity (green) and macrophage accumulation (red). At 20 weeks of age, osteogenic activity was almost undetectable where macrophages accumulated. 10 weeks later in the same lesion of the same mouse, as macrophage burden had increased, marked osteogenic activity was detected. However, 10 weeks of statin treatment in Apoe−/− mice that started at 20 weeks of age suppressed macrophage accumulation and, in parallel, osteogenic activity. (b) Co-mapping of macrophages and calcification by FRI. FRI co-localized macrophage accumulation and osteogenic activity in the aortic root and aorta (Reproduced from Aikawa E. et al. [54])
Following protocol is an example of an intravital molecular imaging using NIRF probes.
Materials
Animals: mouse (Apoe−/−, Ldlr−/− mice, etc.)
Diet: high-fat/high-cholesterol diet (D12108C, Research Diets)
NIRF probes: MMPSense 680, AminoSPARK (PerkinElmer)
Imaging instrument: confocal microscopy (FV1200, Olympus)
Surgical tools: small spring scissors, forceps, polyethylene tubing, corkboard
Anesthetic agents: ketamine, xylazine
Protocol
1.
Prepare model animals (e.g., high-fat diet-fed Ldlr−/− mice).
2.
Intravenously inject NIRF probes.
3.
24 or 48 h later, anesthetize the mice.
4.
Place the mouse on the corkboard at supine position and cut the neck skin at midline.
5.
Separate left and right lobes of thyroid grand by midline incision.
6.
Expose trachea and remove connective/fat tissues around the carotid artery.
7.
Insert polyethylene tubes under the carotid artery to keep the artery horizontal.
8.
Set the mouse on the stage of the microscopy and detect and image the NIRF signals with appropriate wavelength laser.
8.7 Tissue Harvesting
Materials
Mice (high-fat/high-cholesterol diet fed)
Phosphate-buffered saline (PBS) (Cat: 12001-680, VWR)
0.9 % sodium chloride solution (can be used to substitute the PBS above) (Cat: 85583, Owens and Minor)
Surgical scissors, sharp-blunt, one pair (Cat: 14001-12, Fine Science Tools)
Adson forceps, one pair (Cat: 91106-12, Fine Science Tools)
Dumont #5/45 Forceps (angled), two pairs (Cat: 11251-35, Fine Science Tools)
Vannas Spring Scissors, one pair (Cat: 91500-09, Fine Science Tools)
10 mL syringe (Cat: BD309604, VWR)
1 mL syringe (Cat: BD309659, VWR)
25-gauge needles (Cat: 305122, VWR)
Heparin sodium (Cat: 33185, Henry Schein Animal Health)
Pentobarbital (Euthasol) (Cat: 07-805-9296, Patterson Veterinary)
Isoflurane (can be used to substitute pentobarbital above) (Cat: 029405, Henry Schein Animal Health)
Protocol
1.
Either inject pentobarbital (200 mg/kg IP or approved amount for euthanasia) or place the animal under the effect of isoflurane anesthesia.
2.
To first collect whole blood sample for lipid analysis (if blood sampling is not needed, then skip to step 4), hold abdominal skin at midline of the body with Adson forceps and open the abdominal cavity laterally to the diaphragm using surgical scissors—sharp-blunt. Use the forceps (or a cotton swab in substitution) to gently move the digestive tract and other organs to the side to expose the inferior vena cava (IVC) and right kidney.
3.
Coat the inner 1 mL syringe and 25G needle with heparin (drawing as much heparin as you can and flushing it all out). Carefully puncture the inferior vena cava (IVC) and draw blood slowly. A 25–30 g mouse should yield approximately 800–1000 uL of blood collection in volume.
4.
From below the sternum, carefully open the thoracic cavity anteriorly using surgical scissors until the thyroids are exposed. Both sides of the rib cage should be retracted to the side to expose the lungs and heart. Diaphragm can be opened further if more “work space” is needed.
5.
Using micro scissors, snip the right atrium of the heart, and perfuse the remaining blood from the left ventricle using a 10 mL syringe full of PBS or 0.9 % sodium chloride solution.
6.
Remove all other organs and tissues from the body cavity so that only the heart and kidneys are left. Note: take extra caution when removing the lungs, thyroids, and esophagus so that the aorta and carotid arteries are not accidentally damaged. These can be resected using the micro scissors and angled forceps, under a dissecting microscope.
7.
Taking care to not damage the aortic arch, hold surrounding connective tissues with the angled forceps and “peel” away (with micro scissors) as much fatty tissue as possible. Free the left subclavian artery, left common carotid artery, and the brachiocephalic artery (can further free the right subclavian and right carotid artery bifurcation if interested). Transect across these three arteries to leave 2–3 mm attached to the aorta. Separate the heart at the aortic root. The abdominal aorta should also be cleaned of connective tissue and fat, all the way down until the internal/external iliac arteries are exposed. Remove the aorta for tissue embedding.
8.8 Histopathological Approaches
Histopathology is an important tool to characterize pathomorphological changes in human and experimental atherosclerosis. To better understand the development of atherosclerosis and atherosclerotic lesion formation and its composition, it is crucial to know the morphology of the tissue and the various pathological processes involved. Histopathology can also examine the effects of new therapies in vivo. These goals can be accomplished through a variety of histological stains, identification of different proteins through immunohistochemistry, and rigorous quantitative analysis. This section will describe how to process the tissue for histological analyses, the different histological stains used to visualize tissue morphology, and how to identify protein targets through immunohistochemistry.
8.8.1 Embedding of Fresh-Frozen Tissue in Optimum Cutting Temperature (OCT) Compound
Tissue preparation is a critical step in obtaining reliable and reproducible results in histopathology. There are two major types of specimen preparation routinely used in staining experiments: paraffin-embedded and fresh-frozen tissue samples. While paraffin embedding has its advantages (e.g., tissue preservation), fresh-frozen embedding is the preferable method, particularly for immunohistochemistry. Unfixed fresh-frozen tissues maintain antigen preservation. This method also allows more flexibility in post-fixative choice, which can be determined based on a specific experimental goal or the choice of an antibody. This is important because different fixatives (e.g., formalin, paraformaldehyde) may differently affect the reactivity of each antigen. Paraffin embedding of fixed tissues preserves excellent tissue morphology. However, its use for immunohistochemistry may suffer with antigenic masking by a fixative and paraffin embedding (e.g., heat).
One must be extremely careful when freezing tissue. Improper freezing process may cause ice crystal artifacts in the tissue. This often makes morphological interpretation difficult. If tissue is frozen too slowly, large ice crystals can form, producing cracks in the tissue, increasing intracellular and extracellular spaces, the detachment of the tunica intima from the media, and rupturing cell membranes. While “snap freezing” technique using liquid nitrogen allows tissue to be frozen rapidly, tissue often cracks due to the rapid expansion of ice, producing problems later on during sectioning. Liquid nitrogen, in its vapor phase, can also act as an insulator, causing uneven tissue freezing and adding further to ice crystal formation. The ice artifacts due to too slow and too rapid freezing techniques could be overcome by freezing the tissue in chilled/dry ice isopentane (2-methylbutane) (see protocol below). The use of isopentane allows the tissue to freeze without expansion and tissue damage, making it the preferred method for histological and immunohistological applications.
Protocol (Fresh-Frozen Tissue Embedding)
1.
Place a container filled with isopentane (2-methylbutane) onto dry ice 15–20 min before embedding so that the isopentane becomes cold.
2.
Dissect the tissue (approximately 1 × 1 cm).
3.
Incubate the tissue in optimal cutting temperature (OCT) compound (Tissue-Tek O.C.T. Compound, Sakura Finetek) at room temperature (2–3 changes, 1 min each).
4.
Add OCT into a plastic Cryomold.
5.
Place tissue in Cryomold with OCT (make sure that tissue is completely covered by OCT).
6.
Using long forceps, place the Cryomold into cold isopentane until OCT stops bubbling (~1–3 min).
7.
Place frozen mold onto dry ice until isopentane fumes evaporate before going to −80 °C freezer.
Before tissue samples are sectioned, they should be taken out of the −80 °C freezer and placed into the cryostat until the temperature of the tissue falls to the temperature of the cryostat. Sections should be cut in temperatures of around −16 to −22 °C. If cryostat temperature is too low, tissue sections tend to fold during sectioning, making it difficult to get a clean section on the microscope slide. On the other hand, if cryostat temperature is too high, tissue sections will shrivel up during cutting. Therefore, it is crucial to maintain the right cryostat temperature in order to obtain a clean and undamaged tissue section. Furthermore, for immunohistochemical applications, sections are cut in 6–7 μm thickness (thickness can be adjusted based on each experimental situation). Following cryotomy, slides are left at room temperature for up to 30 min to air dry. The contrast in heat melts OCT on the slide, providing better tissue adhesion. The slides are then stored at −20 °C until further needed.
8.8.2 Collagen and Elastin
Elastin and collagen are major extracellular matrix components that influence the progression of atherosclerosis. In response to inflammation, special enzymes are released that degrade elastin and collagen. This leads to the thinning of the fibrous cap, which ultimately leads to its rupture. The following Masson trichrome stain, picrosirius red stain, and van Gieson stain can highlight these extracellular components in atherosclerotic lesions.
8.8.2.1 Picrosirius Red Stain
Picrosirius red stain is an excellent method to detect fibrillar collagen accumulation and tissue collagenous architecture when viewed under circulatory polarized light [16, 51, 59–61]. To accurately quantify fibrillar collagens, assessment with polarized microscopy is essential, while bright-field assessment may overestimate or underestimate the results [62–65]. The use of polarized light also enables to visualize the collagen fiber orientation and perform quantitative analyses on collagen retardation and red and green hues [50, 66] (Fig. 8.2a).
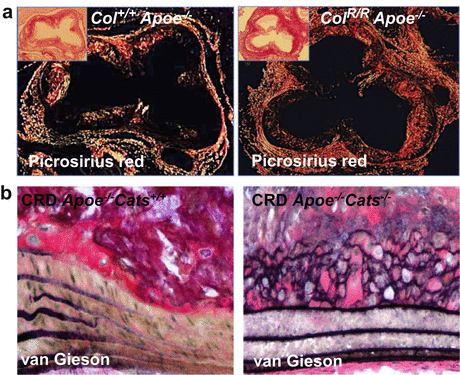
Fig. 8.2
Extracellular matrix staining in mouse atherosclerotic plaques. (a) Picrosirius red staining for fibrillar collagen viewed under polarized microscope [50]. Collagen accumulation in the intima of the aortic root of hyperlipidemic Apoe−/− mice crossed with collagenase-resistant mutant knock-in mice that express type I collagen that is not cleaved by the MMP family collagenases (Col RR /apoE −/−; right) was greater than that of mice expressing wild-type collagen type I (Col +/+ /Apoe −/−; left). Insets: Picrosirius red staining without polarization. (b) van Gieson staining for elastin. Elastin fiber broke in the atherosclerotic plaque of Apoe −/− mice (left). Elastin was better preserved in Apoe −/− mice that lacks cathepsin S (Cats−/−) mouse atheroma [54] (Reproduced from Fukumoto Y. et al. [50] and Aikawa E. et al. [54])
Protocol (Frozen Sections)
1.
Dry slides for 30 min.
2.
Place slides in 10 % neutral buffered formalin for 10 min.
3.
Rinse slides under running tap water for 5 min.
4.
Place slides in distilled water 2 times for 2–3 min.
5.
Incubate slides with 0.1 % picrosirius red (pH = 2) for 90 min.
6.
Rinse in 0.01 N HCL, 2 times for 2 min.
7.
Rinse in distilled water.
8.
Rinse slides in 70 % alcohol for 30 s.
9.
Rinse slides in 95 % alcohol for 30 s.
10.
Rinse slides in 100 % alcohol for 30 s.
11.
Place slides in xylene for 3 min.
12.
Mount slides using Permount.
Results
(a)
Light microscopy
1.
Collagen: Red
(b)
Get Clinical Tree app for offline access

Polarized microscopy
1.
Thick collagen fibers: Yellow, orange
2.




Thin collagen fibers: Green
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree