Fig. 7.1
The SCN from the rat. Top row: Nissl staining at two coronal levels of the suprachiasmatic nuclei (SCN). Middle left: The retino-hypothalamic tract (RHT) as seen by autoradiography to 3H-proline injected to the posterior chamber of the eye. Middle and bottom right: Coronal sections showing immunofluorescence to vasopressin (AVP, rhodamine) and vasoactive intestinal polypeptide (VIP, fluorescein). Bottom left: Double immunohistochemistry to AVP (brown) and VIP (dark blue) from a horizontal slice, caudal (cau) and rostral (ros) are indicated. Other abbreviations: OC optic chiasm, 3V third ventricle, AHA anterior hypothalamic area. Calibration bar: 500 μm top row; 800 μm, middle left, 250 μm, middle and bottom right; 900 μm, bottom left arrow
Fig. 7.2
SCN lesion in hamster abolishes wheel-running circadian rhythmicity, while grafting fetal hypothalamic area containing SCN restores rhythmicity. (a) Reconstruction of electrolytic lesion (dotted area) and location of the graft (diagonal lines) from the same animal showed in b. (b) Actogram of wheel running of a hamster sustaining an SCN lesion 2 weeks earlier. LD light–dark (14:10 h), DD constant darkness, G the day the animal received the fetal SCN graft. SK skeleton photoperiod (1 h lights on, 13:9 h dark intervals). (c) Dark field micrograph from VIP immunostaining of the graft placed in rostral third ventricle (shown in the red rectangle C in a). (d) Dark field micrograph from AVP immunostaining of the graft located caudally in the third ventricle (shown in the red rectangle d in a). (e) Dark field micrograph from adjacent section to d showing retinal ganglion projections to the graft labeled by TMB reaction to cholera toxin injected to the posterior eye chamber. Modified from Aguilar-Roblero et al. (1994)
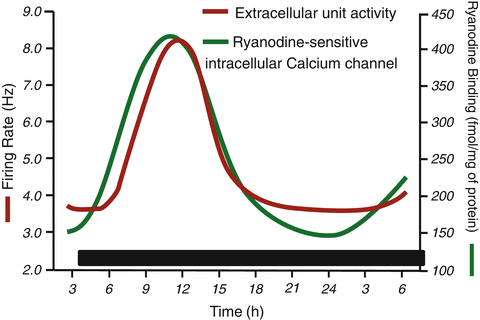
Fig. 7.3
Circadian rhythms in acute SCN slices kept in vitro. Firing rate and ryanodine binding to SCN were obtained in independent experiments. The time correlation of the opening of the ryanodine-receptor intracellular Ca2+ channel (RyR) indicated by increased binding to the rise in electrical activity leads us to suggest a causal relation
7.2 The Molecular Circadian Clock
The molecular clocks in eukaryotes are constituted by transcription–translation feedback loops (TTL) in which dynamics lead them to oscillate with a circadian (≈24 h) periodicity. In Drosophila and rodents, the core genes of the TTL share a promoter region called E-Box in which helix–loop–helix transcription factors bind and initiate gene transcription; in turn, the proteins from these genes accumulate in the cytoplasm and return dimerized to the nucleus, where they dislocate the binding of the helix–loop–helix transcription factors to the E-Box and suppress their own transcription. In rodents the canonical model of molecular circadian clock involves the following processes: the transcription factors CLOCK and BMAL-1 dimerize in the nuclei and bind to E-Boxes in the promoters to induce transcription of the following genes, per1, per2, cry1, and cry2. The proteins translated from the corresponding mRNAs (PER1, PER2, CRY1, and CRY2) are phosphorylated in the cytoplasm and form heterodimers among themselves to enter back into the nucleus. Once in the nucleus, the PER/CRY dimers inhibit their own transcription by removing CLOCK/BMAL1 complex from the E-Box. PER/CRY dimers are eventually removed from the nucleus and hydrolyzed, thus allowing the CLOCK/BMAL complex to bind again to the E-Box, and the cycle reinitiates with a periodicity close to 24 h (Lowrey and Takahashi 2004; Lee et al. 2011). For a recent discussion of the TTL model, see Brown et al. (2012).
The molecular oscillator described above has been demonstrated not only in SCN neurons but also in other brain regions and peripheral tissues (Yamazaki et al. 2000; Balsalobre et al. 2000; Wilsbacher et al. 2002; Reppert and Weaver 2002; Abe et al. 2002; Nishide et al. 2006), which raised the question whether cells other than SCN neurons should be considered also as circadian clocks. These cells are commonly referred to as peripheral oscillators. Transgenic rats and mice that transcribe luciferase—the enzyme responsible to the light emission in fireflies—every time the promoter of the per1 gene is transcribed, have been used as real-time reporters of circadian dynamics. Cultured SCN explants from these animals are able to sustain circadian rhythms in per1-driven luminescence for prolonged intervals of time, whereas in explants from peripheral oscillators, the luminescence rhythms dampen after two to seven cycles (Yamazaki et al. 2000; Wilsbacher et al. 2002; Izumo et al. 2003). These results led to suggest that peripheral oscillators are driven by the central pacemaker in the SCN. This view was challenged by the finding of long-term (up to 20 days) luminescence circadian rhythms in explants from a knockin mouse expressing Period2::Luciferase fusion protein, in both the SCN and peripheral oscillators. These results demonstrate that peripheral tissues also contain self-sustained circadian oscillators that do not depend on the SCN and shift the role of the SCN to a phase coordinator rather than a central pacemaker driving peripheral oscillators (Yoo et al. 2003). Furthermore, the dampening of rhythms found at peripheral tissue level in per1-luc transgenic animals could be at least partially due to the uncoupling of oscillators at cellular level. This is further supported by the observation that bioluminescence rhythms of peripheral oscillators are restored by a serum shock to the cells in culture (Balsalobre et al. 2000; Jung et al. 2003; Izumo et al. 2003). Thus, peripheral oscillators could be phase coordinated (as suggested by Yoo et al. 2003) by the SCN outputs in vivo and probably regulate circadian rhythms at specific tissue and cellular level (Prolo et al. 2005; Loh et al. 2011).
7.3 Beyond the Transcription–Translation Feedback Loop
Previous findings underline the relevance of intercellular communication in the coupling processes among oscillators, which has led to a reevaluation of the role of the SCN neuronal network to maintain coherent phases as part of the clock mechanism. Cultured explants from the SCN present long-term luminescence rhythms with relatively stable phase relations as long as the network communication is intact. Although even in this conditions different regions within the SCN are not completely synchronous to each other, a wave of activation spreading throughout the nuclei with earliest peaking populations of neurons was observed in the medial and dorsal SCN (Yan and Okamura 2002; Yamaguchi et al. 2003; Evans et al. 2011; Foley et al. 2011; Nakamura et al. 2005). In contrast, in cultures from dissociated and dispersed SCN neurons studied at the individual cell level, the rhythms persist for long periods of time, but the phases drift apart from each other. These results indicate that although each SCN neuron has the molecular clock machinery, the SCN network plays a relevant role maintaining a coherent circadian signal by coupling individual oscillators. Further suggested roles of the SCN network are to provide stability and robustness to the circadian clock (Mohawk and Takahashi 2011; see Chap. 9 by Morgado et al.).
Recent studies indicate that the clock mechanism may involve other elements besides the molecular oscillator previously described. The inhibition of adenylyl cyclase by the administration of MDL-12,330A has been shown, in a dose-dependent manner, to decrease the amplitude of circadian transcription and protein synthesis in SCN explants from mPer1::luciferase and mPER2::LUC mice. This clearly indicates that intracellular cAMP signaling is necessary to sustain the progression of the transcriptional rhythms, probably by the activation of CRE sequences (O’Neill et al. 2008). There is also evidence that neuronal firing feedback amplifies the molecular oscillator. In Drosophila electrical silencing of pacemaker neurons disrupts circadian rhythms in locomotion and clock gene expression (Nitabach et al. 2002). In mammals, it has also been shown that action potentials are crucial for a robust expression of molecular rhythms. The blockade of generation of action potentials in the SCN neurons by tetrodotoxin (TTX), a toxin that blocks fast voltage-gated Na+ channels, results in a decay of expression of circadian luminescence in SCN explants from the mPer1-Luc mouse; when analyzed at the cellular level, the amplitude of luminescence rhythmicity decreases and in some neurons disappears, while the individual acrophases among those cells that remain rhythmic become desynchronized (Yamaguchi et al. 2003). The observed effects of TTX on circadian per1-driven luminescence rhythms may depend on Ca2+ influx that results from membrane depolarization during action potentials, since Ca2+ influx also contributes to sustain the molecular circadian oscillation in SCN neurons. Per1-luc rhythmicity from SCN rat explants and PER2::LUC mouse explants is abolished by decreasing Ca2+ concentration in the extracellular medium below 300 mM or by the administration of increasing doses of the intracellular Ca2+ chelator BAPTA-AM (Lundkvist et al. 2005). This hypothesis that Ca2+ influx is necessary to sustain the molecular circadian oscillation is also supported by the elimination of Per2 and Bmal1 circadian expression in cadmium-treated SCN2.2 cells (Sang-Soep et al. 2005).
7.4 Ionic Currents Involved in Neuronal Firing in the SCN
In order to be useful to the rest of the organism, the circadian time signal generated by the molecular clock must be translated into a neuronal firing pattern.
SCN neurons show circadian oscillations in firing frequency in vivo (Inouye and Kawamura 1979) and in vitro (Shibata et al. 1982; Green and Gillette 1982; Welsh et al. 1995); SCN neurons produce action potentials at a higher frequency and show a higher input resistance during the subjective day than during the subjective night. Neuronal firing rate and membrane excitability in SCN neurons depend on the interaction of a number of ionic currents through the plasma membrane (Schaap et al. 2003; Kuhlman and McMahon 2006; Brown and Piggins 2007; Aguilar-Roblero et al. 2009; Colwell 2011). The action potential is triggered by a combination of currents including a slow-inactivating Na+ leak current sensitive to riluzole (Kononenko et al. 2004a, b), a persistent Na+ current insensitive to TTX (Pennartz et al. 1997; Jackson et al. 2004), a hyperpolarization-activated conductance (I h) (Akasu et al. 1993; de Jeu and Pennartz 1997), and a hyperpolarization-activated conductance gated by cAMP (HCN) (Atkinson et al. 2011). The progressive activation of such inward currents drives the membrane potential from the afterhyperpolarization at the end of an action potential to the threshold of activation for the voltage-gated fast Na+ channels, as well as the L- and T-type Ca2+ channels responsible for the next action potential (Huang 1993; Pennartz et al. 2002; Nahm et al. 2005). Repolarization of the membrane is driven by outward K+ currents such as the fast delayed rectifier (fDR) channels and the transient A-type K+ current (I A), which have been demonstrated to modulate the duration of the action potential and thus to participate in the modulation of SCN firing frequency (Bouskila and Dudek 1995; Itri et al. 2005; Kudo et al. 2011). Finally, the afterhyperpolarization is mainly driven by several Ca2+-modulated K+ currents such as the large-conductance current (BK) iberiotoxin sensitive (Pitts et al. 2006; Vandael et al. 2010), the small-conductance current (SK) apamin sensitive (Teshima et al. 2003), an iberiotoxin- and apamin-insensitive K+ (Ca) current (Cloues and Sather 2003), and a barium-sensitive K+ current (K(Ba)) (De Jeu et al. 2002).
7.5 Molecular Clock Control of SCN Neuronal Firing Rate
The circadian modulation of SCN neuronal firing rate is essential for the expression of circadian rhythms. At the time, the mechanisms for the control of membrane excitability by the molecular circadian clock components are not completely identified. Ion channel activity can be either regulated by differential expression of genes coding for membrane channels or, as often found, by posttranslational modifications of these proteins. Outside the SCN, numerous studies have demonstrated the importance of kinase/phosphatase activity in mediating short-term changes in channel function that alter electrical excitability (Smart 1997; Mathie 2007; Okamura 2007; Ebner-Bennatan et al. 2012; He et al. 2013; Kyle et al. 2013).
Circadian modulation of ionic currents has been reported in L-type Ca2+ and K+ conductances, but its contribution to the expression of circadian SCN firing has only been clearly shown for K+ (Colwell 2011). High firing rate from SCN neurons during the day is regulated by fDR K+ currents (Itri et al. 2005). The fast kinetics of fDR K+ currents allows quick repolarization of the neuronal membrane to end the action potential without altering action potential threshold or amplitude (Rudy and McBain 2001). Acute blockade of the fDR with 0.5 mM 4-aminopyridine (AP) or 1 mM tetraethylammonium (TEA) during the day decreases the firing rate of SCN neurons, thus blunting the day/night difference in spontaneous activity, while long-term application of 4-AP (0.5 mM) prevents the expression of multiunit electrical activity rhythm from the SCN (Itri et al. 2005). Lower firing rate of SCN neurons during the night involves the large-conductance Ca2+-activated K+ channel (BK). The activity and mRNA levels of this channel is increased during the night as compared to the day (Pitts et al. 2006). Interestingly, pharmacological block of BK in this study led to a significant reduction of spontaneous spike frequency during the day. This suggests a dual role of BK in regulating spike frequency, which was also found after genetic activation of BK in SCN neurons (Montgomery and Meredith 2012). Kcnma1−/− knockout mice, which lack BK channels, show disrupted behavioral circadian rhythms and a high SCN neuronal action potential frequency during the night, close to the action potential frequency characteristic from daytime recorded SCN neurons, in spite of circadian expression of clock genes such as Bmal-1 and mPer2 (Meredith et al. 2006; Kent and Meredith 2008).
7.6 Intracellular Ca2+ as an Output from the Molecular Clock in SCN Neurons
Neuronal calcium dynamics embodies a code in which the frequency and the amplitude of calcium transients regulate the transcriptional and metabolic status of the cell (Pralong et al. 1994; Spitzer 2002; Neher and Sakaba 2008). Ca2+ homeostasis in neurons involves (1) its influx through voltage- or ligand-gated ionic channels or (2) its release from the endoplasmic reticulum, Golgi apparatus, and nucleus (Finkbeiner and Greenberg 1998), (3) Ca2+ ATPases located in the plasma membrane and endoplasmic reticulum, and (4) several Ca2+-binding proteins (Carafoli et al. 2001). Thus different Ca2+ pools have specific roles in regulating the spatiotemporal neuronal responses to Ca2+ signaling (Rizzuto 2001). Intracellular Ca2+ stores in the endoplasmic reticulum are a key component in the dynamic of [Ca2+]i. Thapsigargin-sensitive Ca2+-ATPase is responsible for the Ca2+ uptake into the stores, while Ca2+ release depends mainly on high-conductance Ca2+ channels termed ryanodine and inositol (1,4,5)-triphosphate receptors, RyRs and IP3Rs, respectively (Meldolesi and Pozzan 1998; Verkhratsky 2005).
In the SCN there are reports on the role of intracellular Ca2+ as a signal involved in resetting the clock, maintaining the molecular oscillation and transmitting the rhythmicity from the clock itself to the efferents involved in the expression of the overt rhythmicity. The manipulation of calmodulin levels phase shifts metabolic and electrical rhythms in SCN neurons (Shibata and Moore 1994 ), while RyRs mediate light-induced phase delays during early subjective night (Ding et al. 1998). The role of Ca2+ influx in sustaining the circadian oscillation of the per1 and per2 clock gene has been shown in SCN neurons (Lundkvist et al. 2005; Sang-Soep et al. 2005).
A circadian rhythm in free cytoplasmic Ca2+, originating from intracellular Ca2+ storages, may be one of the first transmission elements linking the molecular oscillator to the circadian modulation of SCN activity, particularly the modulation of SCN firing rate (Fig. 7.3). In the SCN intracellular Ca2+ regulation shows a clear circadian rhythm. The SCN expresses both types of intracellular Ca2+ channels, inositol triphosphate (IP3) and ryanodine sensitive (RyR). Binding experiments with radioactive ligands demonstrate that RyR but not the IP3 presents circadian regulation in constant darkness, while in other areas of the brain outside the SCN, both IP3 and RyR activity remain constant throughout the day. The peak expression of the RyR binding circadian rhythm in the SCN occurs during the middle of the subjective day at circadian time (CT) 07 and is due to an increase in the protein expression of the neuronal RyR type 2 (RyR2), present in neurons but not astrocytes (Díaz-Muñoz et al. 1999) (Fig. 7.4a-c). Circadian rhythmicity in mRNA expression of RyR is under direct control of bmal1, one of the positive regulators of the clock genes, as bmal1 knockout mice show decreased expression of RyR mRNA and protein (Pfeffer et al. 2009).
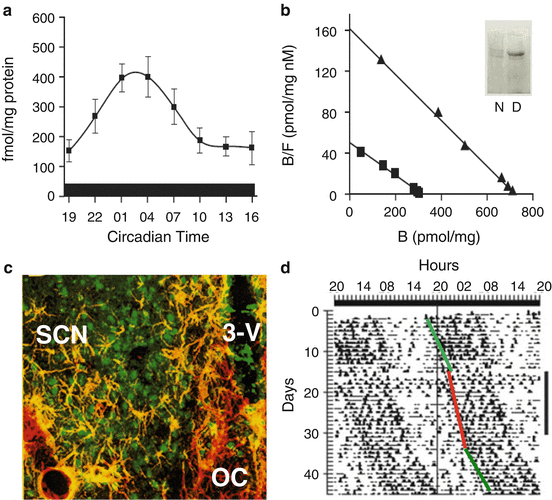
Fig. 7.4
The circadian rhythm in the RyR binding to ryanodine (a) corresponds to changes in Bmax as indicated from Scatchard analysis (b) comparing day (triangles) vs. night (squares); Western blot to RyR type 2 confirmed higher levels of protein during the day (inset). (c) Confocal microscopy showed that RyR-2 immunofluorescence was located in neurons but not in astrocytes (labeled to GFAP). Abbreviations: SCN suprachiasmatic nucleus, 3V third ventricle, OC optic chiasm. (d) Actogram of drinking behavior from a rat receiving 2 weeks of 100 nM ryanodine into the SCN. The drug was continuously delivered (vertical black bar on the right) with an ALZET miniosmotic pump. The period of rhythmicity shortened during the administration (red line) in comparison with before and after the administration (green lines). Since neither the period nor the phase was affected but only ryanodine administration, we conclude the effects were in the output pathway from the clock to the effectors of rhythmicity
Concentrations of free intracellular Ca2+ also show a clear circadian fluctuation as measured with fura2 AM. During the subjective day, (ZT 1–6) [Ca2+]i is higher as compared with the subjective night (ZT 13–18), and this circadian fluctuation occurs even in animals housed in constant darkness (Colwell 2000). This rhythm was abolished with the administration of TTX, which blocks Na+ channels, or methoxyverapamil that blocks L-type Ca2+ channels, concluding that membrane events are associated with this rhythm. In contrast, in SCN organotypic cultures, using a chameleon Ca2+ reporter, circadian rhythms in [Ca2+]i and neuronal firing rate can be dissociated (Ikeda et al. 2003). In this study TTX abolished the rhythm in SCN neuronal firing but did not affect the rhythm in [Ca2+]i. However, closing the intracellular Ca2+ channel sensible to ryanodine (RyR) with high doses of ryanodine abolished both the rhythmicity in intracellular Ca2+ concentration and electrical activity in SCN neurons. In contrast, the inhibition of voltage-gated Ca2+ channels by nifedipine did not affect any of the rhythms.
Ryanodine at a dose of 0.1 μM activates the RyR and causes release of Ca2+ from the endoplasmic reticulum, while ryanodine at a dose of 100 μM has the opposite effect and prevents the release of Ca2+ (Chu et al. 1990). In rats recorded in constant dim red light, chronic administration of different doses of ryanodine into the SCN via a cannula connected to an osmotic minipump has an effect on behavioral circadian rhythms (Mercado et al. 2009). Intracellular Ca2+ release by the activation of RyRs by 0.1 μM ryanodine induced a significant shortening of the endogenous period (Fig. 7.4d), whereas the inhibition of the Ca2+ channels by 100 μM ryanodine disrupted the circadian rhythmicity. After withdrawing the pharmacological treatments, the period and phase of rhythmicity returned to basal and expected values. These results suggest that changes in overt rhythms induced by both doses of ryanodine did not directly involve the clock mechanism.
Pharmacological manipulation of ryanodine-sensitive intracellular Ca2+ channels modifies SCN neuronal firing rate, recorded by perforated patch from rat brain slices (Aguilar-Roblero et al. 2007). Thus, the increase of [Ca2+]i by opening the RyRs by the administration of either 1 mM caffeine or 100 nM ryanodine increases the firing frequency in 58 % of SCN neurons, without having any effect on the firing threshold. Closure of RyRs by the administration of 10 μM dantrolene decreases firing rate in 64 % of the tested neurons, whereas the administration of 80 μM ryanodine decreases firing rate in 100 % of neurons selected by their irregular firing rate and clear hyperpolarizing after potential during basal conditions. Synaptic activity in the SCN can be suppressed by pharmacological blockade of both GABAergic and glutaminergic receptors. Thus, the administration of a cocktail containing 10 μM bicuculline (GABAA receptors), 50 μM AP-V (NMDA receptors), and 10 μM DNQX (AMPA receptors) inhibits synaptic transmission but does not prevent ryanodine effects on firing frequency (increase by 100 nM or decrease by 80 μM), clearly indicating that ryanodine modulation of SCN firing rate is due to a direct action on intracellular Ca2+ mobilization through RyRs and not to indirect effects on the synaptic network (Fig. 7.5). Since the effects of both doses of ryanodine were observed both during daytime (ZT4–10) and nighttime (ZT17–22), the pharmacological gating of the RyRs is able to override the circadian clock control of SCN neuronal firing rate.
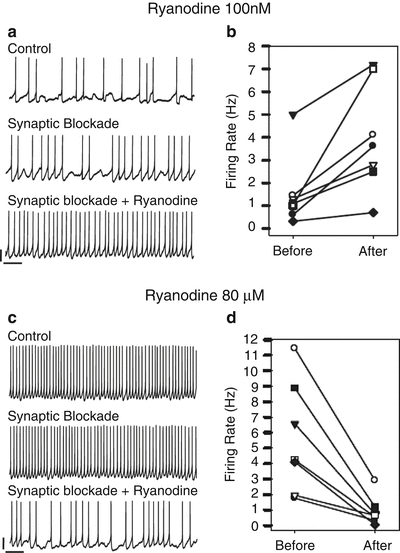
Fig. 7.5
Pharmacological manipulation of the RyRs modulates the firing rate from SCN neurons recorded in perforated patch configuration. Low dose of ryanodine (100 nM) opens the RyR and releases Ca2+ from the endoplasmic reticulum to the cytoplasm, while a high dose (80 μM) closes the RyR and leads to a decrease in free Ca2+ in the cytoplasm. a and c show typical traces from two neurons receiving either 100 nM or 80 μM ryanodine. Synaptic blockade to isolate the recorded neuron from the network was accomplished by a cocktail of bicuculline (10 μM), AP-V (50 μM), and DNQX (10 μM). b and d show the summary of neurons receiving either 100 nM or 80 μM ryanodine. Modified from Aguilar-Roblero et al. (2007)
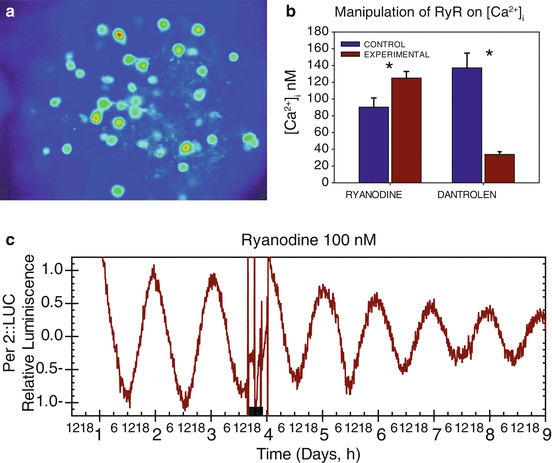
Similar results were found in SCN slices from C57Bl/6 mice. Activating RyRs with 100 nM ryanodine increased [Ca2+]i in about 40 % of neurons, as measured with the Ca2+ indicator dye Fura-2 (Fig. 7.6a). Application of the RyR antagonist dantrolene (10 μM) decreased free cytoplasmic Ca2+ in 54 % of the recorded neurons. The responses to the RyR agonist or antagonist were dependent on the basal [Ca2+]i of the cell (Fig. 7.6b) which may in turn be correlated to the activation state of the RyR. Neither activation of nor blocking of the RyR had major effects on the period or phase of PER2 expression assessed by luminescence in SCN slices from PER2::LUC transgenic mice (Fig. 7.6c). This indicates that acute intracellular Ca2+ release from RyR in SCN neurons does not affect the clock angular velocity (unpublished data). The data are consistent with the behavioral study in rats described above that concluded no impact of RyR activation on molecular clock mechanisms (Mercado et al. 2009). The effects of manipulating the RyR on the neuronal firing, measured in cell-attached configuration, were similar to those previously reported in rats, in which the firing rate increased after 100 nM ryanodine and decreased after 10 μM dantrolene. The proportion of SCN neurons showing ryanodine-induced increase in [Ca2+]i is similar to the percentage of cells increasing neuronal activity after RyR activation. This relationship also holds true for the reduction of [Ca2+]i and spike frequency after blocking the RyR by dantrolene. A potential link between RyR-induced changes in [Ca2+]i and neuronal excitably could be implemented at least in part by the Ca2+-dependent K+ channel BK, which seems to contribute to the high spike frequency during the day (Pitts et al. 2006).