Fig. 7.1.
Synthesis and role of glutathione (GSH). (a) GSH synthesis requires two ATP-dependent enzymatic steps. The first and rate-limiting enzyme is GCL that binds glutamate (Glu) and cysteine (Cys) to form γ-glutamylcysteine (γ-GluCys). GCL consists of two subunits: a heavy catalytic subunit (GCLC) and a light modifier subunit (GCLM). The second enzyme is GSS, which adds glycine (Gly) onto γ-GluCys to form GSH. Here is also depicted the action site of the pharmacological (BSO) and genetic tool (knockout of GCLM) used to induce a GSH deficit. Roles of GSH include antioxidant action and protection against macromolecular damage (b), detoxification (c), and redox-dependent regulation of protein function (d) and DNA binding (2). (b) Antioxidant action of GSH. Hydrogen peroxide (H2O2) and lipid hydroxyperoxide (LOOH) are reduced by GSH via GPX, producing GSSG, which in turn can be reduced back to GSH via GR. Regeneration of ascorbic acid and vitamin E from their oxidized forms (dehydroascorbate and vitamin E radical, respectively) is achieved via the same GPX-dependent mechanism. Failure of this system may result in irreversible macromolecular damage such as lipid peroxidation, formation of protein carbonyls, and DNA damage. (c) Detoxifying action of GSH. Toxic electrophile compounds (Ep), including diethyl maleate and cyclohexene-1-one, are detoxified by formation of adducts with GSH (GS-Ep) via the action of GST, and subsequently expelled from the cells via multidrug-resistance proteins (MRPs depicted as circles). This results in GSH depletion. 4-Hydroxynonenal, a product from lipid peroxidation process, can also be rendered into a non-toxic glutathiyl adduct via a GST-dependent reaction. (d) Redox regulation of protein function. The GSSG/GSH redox couple determines in great part the redox state of cysteine residues on proteins. Reversible oxidation and modification of cysteine residues are important mechanisms that control protein function and interaction between proteins. Oxidation of cysteine residues can lead to formation of disulfide bonds (1) within a same protein (P1) or (2) between two proteins (P1 and P2). The GSSG/GSH redox couple also modulates (3) S-glutathionylation and (4) S-nitrosylation that are known to control protein function. NMDA receptor function is for instance modulated by redox-dependent modifications of cysteine residues located on the NR2A subunit (6). GCL, glutamate cysteine ligase; GPX, glutathione peroxidase; GR, glutathione reductase; GSS, glutathione synthetase; GST, glutathione transferase.
In peripheral tissues of schizophrenic patients, impaired antioxidant defense systems and increased lipid peroxidation have been reported (10, 11). Reductions of plasma antioxidants such as bilirubin and uric acid were found in both first episode (12) and chronic patients (13). Abnormal antioxidant enzyme activity of superoxide dismutase (SOD), GPX, and catalase (CAT) was observed in plasma and/or blood cells of patients (14–17). However, large discrepancies were reported (see (18)). Impaired antioxidant systems could lead to oxidative stress and ROS-mediated injury as supported by increased lipid peroxidation products, decreased levels of membrane polyunsaturated fatty acids (PUFAs), and decreased membrane phospholipids in blood cells (19, 20) and fibroblasts (21). It has been suggested that peripheral membrane anomalies correlate with abnormal central phospholipid metabolism in first episode and chronic schizophrenic patients (22–24).
Altered redox systems have also been observed in brains of subjects suffering from schizophrenia. Decreased levels of GSH in cerebrospinal fluid (CSF) and in medial prefrontal cortex (PFC) have been measured in schizophrenic patients (25). A microarray and proteomic study showed anomalies of mitochondrial function and oxidative stress pathways in post-mortem brains of patients (26). In addition, lower GPX and glutathione reductase activities (27), elevated Mn-SOD protein levels and activity (28, 29), and elevated Cu- and Zn-SOD activity (29) were found in post-mortem brains of patients. However, it was not clear whether these observations indicated an intrinsic impairment of some antioxidant systems or whether these were the consequences of other dysfunctions and/or environmental insults (i.e., toxic compounds, infections) leading to an excess of ROS production.
We recently provided genetic and functional evidence for an intrinsically impaired function of the glutamate cysteine ligase (GCL, Fig. 7.1a), the rate-limiting enzyme of GSH synthesis (30). Polymorphisms of GAG trinucleotide repeat (GAG-TNR) of the catalytic subunit of GCL (GCLC) were associated with the illness in two independent cohorts. Compared to skin fibroblasts of “low-risk” GCLC genotypes, fibroblasts of “high-risk” GCLC genotypes, present in 35–40% of patients, had lower GCL protein expression, GCL activity, and GSH levels under oxidative stress. This demonstrated that GAG-TNR variants are associated with dysfunctional regulation of GSH synthesis in a sub-population of patients. Furthermore, “high-risk” genotype patients had lower plasma GSH levels and higher oxidized cysteine levels than “low-risk” patients, pointing to a generalized redox dysregulation (31). This is consistent with the decreased GSH levels reported in schizophrenia (25, 27). The drug-naïve status of the patients in the CSF study (25) suggested that the deficit in GSH was not a consequence of treatment. However, GAG-TNR polymorphism was also associated with bipolar patients, but not with major depression (Gysin et al., unpublished), supporting the concept of a psychosis continuum (32) and that various genetic anomalies are common to several forms of psychosis. The implication of redox dysregulation in both schizophrenia and bipolar disease is consistent with the observation that supplementation of N-acetyl-cysteine, a cysteine prodrug, improves both schizophrenic and bipolar patients (33–35). Altogether, this suggests that a dysfunctional regulation of GSH synthesis and a subsequent deficit of GSH in brain and peripheral tissue can be a vulnerability factor to schizophrenia, but also possibly to bipolar disease. Furthermore, other susceptibility genes for schizophrenia may also induce an oxidative state. For instance, functional polymorphisms of PRODH, encoding proline oxidase, that are positively associated with schizophrenia display increased enzyme activity (36), leading to subsequent decreased proline levels. Proline has antioxidant properties and protects the intracellular GSH pool and the GSSG/GSH redox status (37). Consequently, low levels of proline could cause redox dysregulation, which could be particularly severe when combined with impaired GSH synthesis.
It is quite striking that many environmental risk factors for schizophrenia (malnutrition, exposure to toxins, maternal infection and diabetes, obstetrical complications, maternal or early life stress, or later insults like brain trauma and stress during childhood, adolescence, and adulthood) result in increased ROS generation, lipid, protein, and DNA oxidation, and decreased GSH levels and antioxidant defense system (see (38) for review). Some of these environmental insults also lead to increased inflammation, emphasizing the tight link between inflammation and oxidative stress (39, 40). Any of the aforementioned environmental insults could worsen the fragile redox equilibrium and, depending on the phase of brain development when they occur, could perturb normal brain maturation with delayed functional consequences in early adulthood. Thus, a genetic defect that leads to dysregulation of GSH synthesis, combined with environmental insults or other genetic factors generating oxidative stress, could be a critical factor contributing to the emergence of schizophrenia (38).
Since the brain is highly vulnerable to oxidative damage because of its high oxygen consumption, its high content of oxidizable polyunsaturated fatty acids, and the presence of redox-active metals (Cu and Fe), a GSH deficit could be particularly damaging to the neuronal function. Oxidative stress-induced cellular damage is also involved in the pathogenesis of various neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (ALZ), and Huntington’s disease (HD). However, in contrast to schizophrenia where a genetic origin of GSH synthesis could affect brain development, GSH depletion and ROS/RNS increase in neurodegenerative diseases appear later in life and are probably downstream consequences of other primary causes (1).
In the following, we review animal models of GSH deficit that have been used to determine the consequences of redox dysregulation and to assess their involvement in the pathology of schizophrenia. The chapter is focused on pharmacological and genetic approaches that induce a GSH deficit in rodents via a downregulation of GCL, the key synthesizing enzyme of GSH. In rodent models, GSH deficit was induced (1) semi-chronically during postnatal development, (2) acutely during adulthood (pharmacological treatment with l-buthionine-SR-sulfoximine, L-BSO), or (3) chronically throughout the life [genetic model GCLM (−/−) mice] (Fig. 7.2). These models were used to address the questions below.
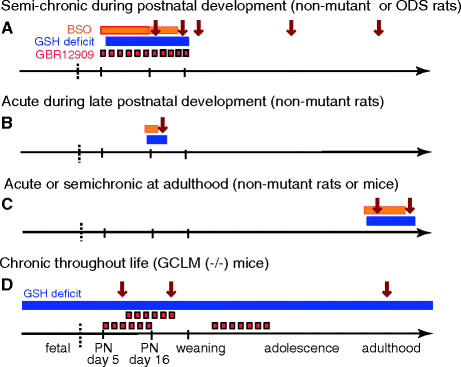
Fig. 7.2.
Schema of the different types of induced GSH deficits that are presented in the review. Horizontal bars delineate the time of the different BSO treatments and the approximate corresponding periods during which a brain GSH deficit is observed. Dotted horizontal bars indicate the period of GBR12909 treatment causing increased extracellular dopamine levels in brain regions rich in dopamine innervations. Arrows indicate times of morphological, physiological, or behavioral analyses. Dotted vertical line indicates birth.
First, what are the short- and long-term consequences of a GSH deficit restricted to a period of the development? This can test the contribution of a redox dysregulation in the neurodevelopmental aspect of schizophrenia. Neuroanatomical, neurochemical, neurophysiological, and psychopathological data converge to suggest that interactions between susceptibility genes and environmental insults during pre- and perinatal development cause defects in neuronal integrity and connectivity, setting off a cascade of events that extend into adult life and lead to the emergence of the psychotic symptoms. In this context, a genetic and/or environmentally derived GSH deficit could represent an important susceptibility factor (see (38) for review).
Second, what are the effects of an acute and transient GSH deficit during adulthood? GSH deficits have been observed in adult patients (25). This approach is useful to distinguish the consequences of an acute GSH deficit during adulthood from those caused by a GSH deficit during development.
Finally, what are the effects of a chronic GSH deficit throughout life? This is expected to combine effects of a GSH deficit during development (including during fetal development) and later in life. However, a chronic GSH deficiency may in addition lead to more profound adaptations of the entire system to compensate for the redox dysregulation.
2 Pharmacological Models of GSH Deficit
Two main types of pharmacological agents are available to induce a GSH deficit: electrophile compounds and specific inhibitors of GCL. Electrophile compounds (i.e., diethyl maleate and 2-cyclohexen-1-one) cause very rapid and reversible depletion of GSH through a detoxifying process. These compounds conjugate with GSH directly or via the GSH S-transferases (41). The resulting conjugates are then expelled from the cells through pumps called multidrug-resistance proteins (MRPs), leading to rapid decrease in intracellular GSH (Fig. 7.1c). These GSH-depleting agents cross the blood–brain barrier and are able to efficiently decrease brain GSH levels within hours, leading to increased protein carbonyls (42, 43). However, they are quite toxic, not very specific for GSH depletion (at least diethyl maleate), and cannot be used for long-term GSH depletion. In our view, they are not appropriate pharmacological tools to model GSH deficit in the context of schizophrenia but are adequate for toxicological studies. However, two studies describe the effects of these GSH-depleting agents on rodent behavior. Adult rats and mice with acute GSH depletion induced by 2-cyclohexene-1-one (for a few hours) showed impaired short-term spatial memory in a Y maze (44). Rats with an acute GSH deficit induced by diethyl maleate (for a few hours) had impaired acquisition of spatial reference memory in a water maze and displayed normal acquisition in an avoidance test (45). Impaired short-term spatial memory in 2-cyclohexene-1-one-treated animals was also reported for other models of GSH deficit (see following paragraphs). However, impairments in spatial learning and memory in a water maze have not been observed in other models of GSH deficit than diethyl maleate-treated rats, suggesting that some effects of diethyl maleate may be independent of GSH depletion.
More specific pharmacological tools are inhibitors of GCL. The most specific inhibitor is dl-buthionine-(SR)-sulfoximine (BSO) (see Fig. 7.1a). It represents a relevant model of GSH deficit, as observed in schizophrenia. Only the l-buthionine-(SR)-sulfoximine (l-BSO) is a mechanism-based inhibitor subject to ATP-dependent, enzyme-catalyzed phosphorylation by GCL to form l-buthionine-(SR)-sulfoximine phosphate which tightly binds to the active site of GCL (46–48). l-BSO is only weakly metabolized as 90% is expelled from the body in its original form. l-BSO is considered to be an irreversible inhibitor. But after a washout period, GSH levels in cells/tissue slowly recover via turnover of GCLC and possibly very slow reactivation (48). The main limitation for the use of l-BSO in vivo is its poor ability to cross the blood–brain barrier after weaning age (49, 50). Therefore, only a transitory GSH deficit during postnatal development can be achieved by subcutaneous (s.c.) injections of l-BSO. BSO s.c. injection in pre-weanling animals (Fig. 7.2a) can be used to investigate the effects of a transitory GSH deficit on postnatal development and its long-term effects in adulthood when GSH levels have returned back to normal. Indeed, GSH levels normalize a few days after the last BSO treatment (51, 52). On the other hand, acute GSH deficit in adults, via intracerebroventricular (i.c.v.) administrations of l-BSO (Fig. 7.2c), can help understand the effects of an acute or a semi-chronic GSH deficit in adulthood, independent of any developmental contribution.
In rats and mice, a GSH deficit causes upregulation of the synthesis of the antioxidant ascorbic acid (53). Such compensation does not exist in humans, as we are not able to synthesize ascorbic acid. In order to use a rodent model that resembles more closely the conditions found in humans, GSH deficit was induced in osteogenic-disorder Shionogi (ODS) rats (54), which are deficient in l-gulonolactone oxidase, the key enzyme in the biosynthesis of ascorbic acid (55). In this rat strain, a GSH deficit does not lead to a compensatory increase in ascorbic acid.
Animals with a GSH deficit can be more vulnerable to any insults that cause oxidative stress. For instance, enhanced dopamine release, as observed during the encounter of a stressful situation (56), could be deleterious and exacerbate redox dysregulation during conditions of GSH deficit. Indeed, catecholamines such as dopamine produce ROS (57) that need to be neutralized by antioxidant systems. These catecholamines can also react with cysteine residues of GSH and proteins to form conjugates, leading to decrease of endogenous GSH (58–60). To test the effect of enhanced dopamine release during a transient GSH deficit during development, a specific inhibitor of the dopamine re-uptake transporter 1-(2-(bis-(4-fluorophenyl)methoxy)ethyl)-4-(3-phenylpropyl)piperazine dihydrochloride (GBR12909) was used to elevate extracellular dopamine levels locally in brain regions rich in dopamine innervations (61).
2.1 BSO-Induced Transient Postnatal GSH Deficit
2.1.1 Methods
Animals
BSO-induced GSH deficits (Fig. 7.2a, b) were studied in non-mutant rats (OFA, Wistars) and in osteogenic-disorder Shionogi (ODS) rats. A GSH deficit leads to compensatory increase in ascorbic acid synthesis in non-mutant rats, but not in ODS rats. ODS rats were obtained from Clea Japan, Inc. (Tokyo, Japan). Because ODS rats cannot synthesize their own ascorbic acid, they require freshly dissolved ascorbic acid (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland) in their drinking water (1 g/l) to prevent vitamin C deficiency (62). ODS rats treated with BSO were in a state of GSH deficiency that was not compensated by ascorbic acid synthesis, but they were not in a state of combined GSH and vitamin C deficiency.
BSO Treatment
A transient postnatal GSH deficit [from postnatal (PN) days 5–24] by about 50% was achieved in brain of rodents through daily subcutaneous (s.c.) injection of l-buthionine-(S,R)-sulfoximine (l-BSO; 3.8 mmol/kg animal corresponding to 0.842 mg/g animal) (51, 63). l-BSO (>99% purity) in powdered form can be purchased from Acros Organics (Geel, Belgium) and stored at 4°C. BSO was dissolved in a phosphate buffer solution (PBS) consisting of 25 mM NaH2PO4, 65 mM Na2HPO4, and 0.9% NaCl at pH 7.4. Other investigators used 0.9% physiological saline (NaCl alone) to dissolve BSO (64). Solubility of BSO was facilitated by sonication and/or heating the solution to about 45°C for 10–15 min. According to the producer, BSO solutions can be stored at –20°C for up to 3 months. Typically for a rat, 100 μl of a freshly made BSO solution (2–4 mM) was injected daily in the morning. Prior to injection, each animal was weighed in order to adjust the BSO concentration to inject (0.842 mg/g animal). This protocol can be adapted to mice.
Enhanced Extracellular Dopamine Levels by GBR12909 Treatment
The specific inhibitor of the dopamine re-uptake transporter, 1-(2-(bis-(4-fluorophenyl)methoxy)ethyl)-4-(3-phenylpropyl)piperazine dihydrochloride (GBR12909), was used to elevate extracellular dopamine levels locally in brain regions rich in dopamine innervations. GBR12909 dihydrochloride (purity >98%) was initially purchased from Tocris Cookson Ltd (Bristol, UK) and stored dessicated at RT. Recently, however, GBR12909 is produced by Biotrend AG (Zurich, Switzerland), on behalf of Tocris. GBR12909 (GBR) was freshly dissolved in PBS (pH 7.4) and subsequently injected s.c. (5 mg/kg animal, in 100 μl) to each rat every other day alone or in combination with BSO (51, 65). Prior to injection, each animal was weighed in order to adjust the GBR12909 concentration to inject (5 mg/kg animal). This protocol can be adapted to mice.
2.1.2 Results
Effects on Brain GSH Levels, Oxidative Stress, and Physical Parameters
While Slivka et al. (50) reported ∼80% GSH depletion in brain of preweaned mice after four daily injections (s.c.) of BSO, a single daily injection (s.c.) of similar BSO doses in rats from PN days 5–16 resulted in only 40–50% diminution of brain GSH (63). Comparable brain GSH deficit was also observed when BSO was daily administrated to 15-day-old rats for only 1–2 days (66); GSH levels returned back to normal within 2–3 days after the end of the treatment (51). BSO-induced postnatal GSH deficit did not affect the activity of the redox-related enzymes CAT, SOD, GPX, and GR (63).
BSO treatment during postnatal development induced similar GSH deficit in ODS and non-mutant rats (63). However, lipid peroxidation was more prominent in ODS rats, particularly in the diencephalon and pons/medulla region (63). Generally, the effects of BSO treatment were stronger in ODS than in any of the non-mutant rats. After 6 days of BSO treatment, ODS rats showed lower body weight than their untreated counterparts (65), but this body weight deficit slowly disappeared after the end of the treatment. Finally, BSO induced more severe cataracts in ODS than in non-mutant rats (65). GSH is indeed an essential antioxidant vital for the maintenance of the transparency of the lenses (67), which are highly vulnerable to oxidative stress. Cataractogenesis due to BSO treatment during postnatal development was largely prevented by melatonin, which has antioxidant properties (68). Altogether, this indicates that the inability to synthesize ascorbic acid (like in humans) is an additional vulnerability factor to a GSH deficiency.
In some experiments, rats with a transient GSH deficit during postnatal development were further challenged by a co-treatment with the dopamine uptake inhibitor, GBR12909 (GBR). GBR causes excess extracellular dopamine, which could further disturb the delicate redox balance. Overall, GBR+BSO-treated rats were not notably different from BSO-treated rats in terms of GSH deficit, lipid peroxidation, body weight, and cataract occurrence (51, 65). However, the additional GBR treatment exacerbated some adverse effects of GSH deficit on the development of cortical circuitry and cognitive functions (see below).
Effects During the Development of Cortical Circuits
The neuronal circuitry of the PFC is altered in schizophrenic patients (for review, see (69)). This includes reduced spines on pyramidal neurons and reduced expression of parvalbumin (PV), GAD67, and GAT1 in fast-spiking interneurons. ODS rats were used to examine whether a transient BSO-induced GSH deficit during postnatal development (with or without GBR-induced elevated extracellular dopamine) could alter cortical circuits in the PFC. In these experiments, rats were sacrificed after the end of the postnatal treatment (at PN day 16 or 24) and brains were processed for either immunohistochemistry (for interneuron investigations) or Golgi preparations (for dendritic spine quantification). BSO treatment alone affected parvalbumin-immunoreactive (PV-IR) interneurons in the anterior cingulate cortex as demonstrated by a reduced number of PV-IR processes radiating from the somata (70). This effect was more prominent in rats treated with BSO+GBR. Moreover, a GSH deficit combined with elevated extracellular dopamine during postnatal development led to a drastic reduction in the number of small PV-IR profiles (corresponding to PV-IR synaptic boutons and dendritic and/or axonal arborization) in superficial (II–III) and deeper (V–VI) layers of the anterior cingulate cortex, but not in the somatosensory cortex (70). However, there was no loss of PV-IR interneurons as the density of large PV-IR (cell bodies) was not altered. The effect of a GSH deficit on PV-IR interneurons was quite specific seeing that expression of other calcium-binding proteins (i.e., calbindin and calretinin) was not significantly altered by BSO or BSO+GBR treatments. In addition, BSO+GBR treatment during postnatal development (PN days 5–24) also altered the morphology of the dendritic spines on apical and basal dendrites of pyramidal neurons of the layer III of the anterior cingulate cortex (Gheorghita et al., unpublished). Altogether, the use of BSO treatment in rats during their postnatal development, particularly when combined with elevated extracellular dopamine, affects cortical circuitry in the prefrontal cortex (i.e., PV fast-spiking interneurons and dendritic arborization of pyramidal neurons). These observations are analogous to anomalies found in post-mortem brains of schizophrenic patients (71–73).
Long-Term Effects on Cortical Circuits
Another important question is whether anomalies in PV-IR interneurons caused by a transitory GSH deficit during postnatal development persist throughout adulthood, as GSH levels normalize after the end of the treatment. This has not yet been fully examined. Preliminary observations indicate that BSO or BSO+GBR treatments during postnatal development lead to reduced number of PV-IR interneurons in the anterior cingulate cortex of adult ODS rats (PN day 90) (Cabungcal et al., unpublished). This effect was less severe after BSO treatment alone. Thus, the alteration of PV-IR interneurons by a transitory GSH deficit during development rendered these interneurons more vulnerable during adulthood. This suggests that a transient GSH deficit during postnatal development is sufficient to affect permanently PV-IR interneurons in the anterior cingulate cortex, including a possible cell loss. This long-term effect on cortical circuitry and possibly other regions could be related to the observed long-term cognitive impairments described below.
Long-Term Effects on Behavior
A general investigation of the neurological status of the rats up to PN day 50 indicated no obvious effect of BSO treatment. Forelimb muscle strength, novelty reaction, escape, and startle reactions were not affected by BSO treatment with or without GBR (51). A transitory GSH deficit during postnatal development impaired neither spontaneous alternation task performance nor exploratory behavior in an open field (51). The main effect in juvenile ODS rats but not in non-mutant Wistar rats (PN days 25–31) was a significant impaired performance in the spatial water maze task that was observed soon after BSO treatment cessation (74). However, this impairment was no more present in older ODS rats (75).
Nevertheless, significant cognitive deficits appeared during the third month of postnatal life. Adult ODS but not non-mutant rats with a transient GSH deficit during postnatal development showed impaired object recognition. This deficit was detected when interval delay was 30 min but not 15 min, suggesting a specific retention impairment. Interestingly, compared to control subjects, the object recognition capacity of schizophrenic patients is also impaired (76–79). The object recognition impairment after BSO treatment emerged earlier in male rats (PN day 65) than in females (PN day 94) and was more severe in males. At PN day 94, males were impaired after BSO treatment (± GBR), while females were impaired only after BSO+GBR treatment (65). This could be due to the protective and antioxidant effects of estrogens (see (80)). Interestingly in humans, first psychotic symptoms occur earlier in men than in women.
BSO-treated rats trained at a later adult age (above 5 months) in classical spatial tasks expressed a severe impairment in a homing board task, but not in a reference and a working memory paradigm in water maze (74, 75, 81). On a homing board, both adult ODS and Wistar rats treated with BSO during postnatal development were highly inaccurate in discriminating the escape hole position based on distributed visual and olfactory cues (74). Their accuracy was restored only when a single salient cue marked the trained position. In a radial maze, adult BSO-treated rats did more working memory errors and were markedly disturbed by the addition of eight different olfactory cues, a procedure that normally enhances the performance of control rats (74). This suggests that BSO-treated rats have difficulties integrating multi-sensory cues. The fact, that performance of adult BSO-treated rats was intact in water maze but impaired in homing board, suggests that either the heavy path integration requirement during successive locomotion bouts on a solid floor or the presence of multiple olfactory cues, or a combination of both factors, enhanced task difficulty in the homing board for the BSO-treated rats. Interestingly, schizophrenic patients show impaired multi-sensory processing (82). The data also revealed that the effects of a GSH deficit were more severe in ODS rats unable to synthesize ascorbic acid or when combined with an additional stress factor (GBR treatment). Altogether, these studies demonstrate that a transitory GSH deficit during postnatal development has long-term behavioral consequences, affecting particularly the processing and integration of multi-sensory cues.
2.2 BSO-Induced Acute GSH Deficit in Adulthood
2.2.1 Methods
GSH deficit in adult rodents was achieved via administration of BSO into the lateral ventricle (Fig. 7.2c). Unilateral injection appears to be sufficient to induce similar GSH deficit contra- and ipsilaterally to the injection site (52). In adult rats, a dose of 3.2 mg BSO/animal (dissolved in 30 μl of physiological saline, pH 7.4) was routinely administrated every 24 or 48 h for the longest treatments (12 days) (83, 84). A single BSO administration corresponds to approximately 0.01 mg/g rat. In mice, daily doses of about 0.02 mg/g mouse (dissolved in 3–5 μl of physiological saline, pH 7.4) were used (52, 64). Below is the description of the intracerebroventricular injection protocol used by Jacobsen et al. (52) on mice, which is easily adaptable to rats or other rodents. Mice were anesthetized using a saline solution (10 ml/kg; i.p.) of ketamine (100 mg/kg) and xylazine (10 mg/kg) and placed in a stereotaxic frame. A guide cannula (cat. no. C313GS-5; Plastics One, Roanoke, VA, USA) was implanted into the lateral ventricle (AP −0.3 mm; ML 0.9 mm; DV 2.0 mm) and fixed in place with two anchor screws (CMA, North Chelmsford, MA, USA) and dental cement. The guide cannula was protected with a dummy cannula (cat. no. C313DC; Plastics One) until used. Mice recovered for at least 1 week before injection and were treated with antibiotics (1.2 mg sulfamethoxazole/ml and 0.24 mg trimethoprim/ml) and analgesics (acetylsalicylic acid; 1 mg/ml) in the drinking water for the first 4 days after surgery and thereafter with only antibiotics. The internal cannula (cat. no. C313I; Plastics One) was inserted into the guide; l-BSO (0.5 mg/mouse; 5 μl) or saline was injected (1 μl/min, 5 min) using a A-99 Razel syringe pump (Stamford, CT, USA). The internal cannula was left in place for 10 min after injection to allow for dispersion away from the injection site.
2.2.2 Results
Effects on Brain GSH Levels, Oxidative Stress, and Physical Parameters
Intracerebroventricular injections of BSO in adult rodents caused a 40–80% decrease in brain GSH levels. All brain structures, even the most distant from the site of injection, were equally affected (52). However, Shukitt-Hale et al. (84) found that after 12 days of treatment, GSH deficit varied in brain regions, with striatum being the most affected and cerebellum the least. Because of the limited number of studies available, it is difficult to know to what extent GSH deficit varies with the animal model, the dose of BSO, and the duration of the treatment. BSO appears to induce faster GSH depletion in mice (52, 64) than in rats (83), but this difference might well be due to the difference in the BSO concentrations used. Maximum GSH deficiency was reached after 48 h in rats (83, 84) and 24 h in mice (52). The degree of GSH deficit remained stable for 12 days following BSO administration every other day. At the end of BSO treatment, brain GSH levels slowly recovered and normalized almost completely after 72 h (52). This decrease in GSH was accompanied by increased protein oxidation (64).
Long-term BSO treatment (12 days) resulted in loss of body weight in rats but did not cause major pathology and did not affect locomotor coordination and performance (84). However, some rats with a GSH deficit exhibited slight and occasional tremors/convulsions, usually in the first few days of BSO treatment (84). Furthermore, Abe et al. (85) showed that BSO-induced GSH deficit in adult mice exacerbated the convulsive action of pentylenetetrazol. This suggests that a GSH deficit can promote epileptic conditions, possibly by depressing the GABAergic system.
Effects on Behavior
BSO-induced GSH deficit in adult rodents did not affect locomotor activity (52), spatial reference learning, and spatial reference memory in a water maze (86). However, when GSH deficit was combined with intraventricular injection of dopamine, impairment of locomotion coordination and spatial learning and memory in water maze was observed (84, 86). Since dopamine alone or injected before BSO injection did not have any effect, the authors suggested that reactive compounds resulting from dopamine autooxidation led to further redox dysregulation in rats with GSH deficit, thus causing impairment in locomotion coordination and spatial performance.
Adult mice with a BSO-induced GSH deficit had reduced capacity in novel object recognition and in a spontaneous alternation task, suggesting impaired short-term memory (64). Interestingly, adult rats which had a transient GSH deficit during postnatal development also showed impaired novel object recognition but not spontaneous alternation (51, 65), suggesting that recognition of objects is particularly vulnerable to a GSH deficit, independent of its time of occurrence. Chronic GSH deficit in GCLM (−/−) mice also led to impaired object recognition (see below).
Finally, a GSH deficit in adult mice led to altered psychostimulant-induced locomotor activity (52). Locomotor activity was only transiently increased by amphetamine in BSO-treated mice, while PCP-induced increase in locomotor activity was strongly enhanced in BSO-treated compared to control mice. Dopamine release induced by amphetamine was enhanced in nucleus accumbens but not in PFC of BSO-treated mice, while PCP-induced dopamine release in nucleus accumbens was not affected by a GSH deficit (52). The behavioral responses of the BSO-treated mice to amphetamine and PCP could not however be simply explained by the effects of GSH deficit on dopamine release induced by these psychostimulants. Responses to psychostimulants depend on several neurotransmission systems (i.e., dopaminergic, serotonergic, noradrenergic, and glutamatergic) and interactions between different brain regions (i.e., striatum, nucleus accumbens, PFC, and VTA). A GSH deficit or the resulting increase in ROS could affect many of the above neurotransmission systems in a complex manner.
Effects on Neurotransmission
Many proteins are redox sensitive and their functions modulated by the redox state of the cell. This includes NMDA receptors (6), GABAA receptors (87), IP3 receptors (88), ryanodine receptors (89), calcineurin (90), various calcium and potassium channels (91, 92), and dopamine and glutamate transporters (93, 94). Thus, a GSH deficit could potentially alter many aspects of neurotransmission.
NMDA receptors. For instance, NMDA receptor-mediated field excitatory postsynaptic potentials (fEPSPs) were weaker in hippocampal slices of BSO-treated rats compared to control rats (66). This hypofunction of NMDA receptors was partly due to an excessive oxidation of their extracellular redox-sensitive sites. In addition, a GSH deficit caused reduced NMDA receptor-dependent, long-term potentiation, decreased paired-pulse facilitation, and increased excitability of pyramidal neurons in CA1 (66).
Dopamine signaling. A BSO-induced GSH deficit also altered dopamine modulation of NMDA-mediated calcium responses in cultured cortical neurons (95). Dopamine decreased NMDA responses in BSO-treated neurons, while the same dopamine concentration (1 μM) enhanced NMDA responses in neurons with normal GSH. The effect of a GSH deficit on dopamine modulation of calcium responses was due to alteration of dopamine modulation of L-type calcium channels and could be prevented by sulpiride, an antagonist of D2-type receptors. This suggests that antipsychotics with D2 antagonist properties might prevent the alteration of dopamine modulation of calcium responses induced by a GSH deficit. Thus, a GSH deficit can lead to changes in neurotransmission and neuromodulation that are relevant to schizophrenia.
3 Genetic Models of GSH Deficit
Both the catalytic (GCLC) and the modifier (GCLM) subunits of GCL, the key enzyme of GSH synthesis, have been associated with schizophrenia (30, 96). In particular, some polymorphisms of GCLC that display blunted oxidative stress-induced increase in GCL activity and GSH levels confer a higher risk to schizophrenia. Therefore, genetic animal models targeting GCL to disrupt GSH synthesis (97) are valuable models to investigate the effects of a GSH deficit in the context of schizophrenia. Knockout of the GCLC gene, however, is lethal at an early embryonic stage (5, 98), presumably because GSH synthesis is completely suppressed. By contrast, GCLM (−/−) mice are viable showing a decrease in GSH of 50–80% depending on the organs (99, 100).
3.1 GCLM Knockout (GCLM −/−) Mice
Association between the GCLM gene and schizophrenia (association with SNPs situated on the 5′ and 3′ regions) was found in two independent cohorts of patients (Swiss and Danish) and in a family-based study from NIMH cohort (96), but not in the Japanese population (101). In the Swiss and Danish cohorts, no clear causal variant within the coding regions of the GCLM gene was however identified (102). However, reduced expression of GCLM mRNA was observed in cultured fibroblasts of patients compared to control in the Swiss sample (96). Therefore, these findings do not rule out the possibility that DNA change, critical for GCLM function or expression, is located in a region that was not screened (e.g., intronic region). Thus, GCLM (−/−) mice (99) represent a valid genetic animal model to study the effects of a chronic and systemic GSH deficit (Fig. 7.2d). GCLM (−/−) mice show about 60% decrease in GSH across brain regions and throughout life (103). GCLM (−/−) mice have no overt phenotype besides a slight reduction in body weight (99) and a reduced female fertility, and cultured cells from these mice are vulnerable to neurotoxicity and ROS (99, 100, 104). However, more detailed investigations revealed that GCLM (−/−) mice display brain morphological, physiological, and behavioral anomalies that could be related to schizophrenia and/or other psychiatric disorders.
3.1.1 Methods
GCLM (−/−) mice were kindly provided by Tim Dalton and Chen Ying from the Department of Environmental Health, Center for Environmental Genetics, University of Cincinnati Medical Center (USA). See (99) for a more detailed description of the generation of these mice. GCLM (−/−) mice were then backcrossed with C57Bl/6 J mice over >10 generations. GCLM (−/−) mice were compared with (+/+) littermates.
3.1.2 Results
Morphological Anomalies
GABAergic system. Compared to (+/+) mice, GCLM (−/−) mice showed selective and region-specific anomalies in the GABAergic system. Like in the BSO-treated rats, PV-IR interneurons in GCLM (−/−) mice were particularly affected. The developmental expression of PV was impaired in both the anterior cingulate and the somatosensory cortex at PN day 10, but normalized at PN day 20 (105). Additional stress induced by GBR treatment during postnatal development of GCLM (−/−) mice (from PN days 10–20) led to reduced number of PV-IR interneurons in the anterior cingulate but not somatosensory cortex until PN day 20 (105). PV-IR interneurons in the anterior cingulate cortex of GCLM (−/−) mice remained vulnerable after weaning, as GBR treatment between PN days 30–40 also caused a decrease in PV-IR interneurons in GCLM (−/−) but not (+/+) mice (Cabungcal, unpublished). Interestingly, elevated oxidative stress, as revealed by 8-oxo-dG (marker of DNA oxidation), was present in the anterior cingulate, but not in the somatosensory cortex of GCLM (−/−) mice at PN day 20 (105) (Cabungcal et al., unpublished). This coincides with the higher vulnerability of PV-IR interneurons in the anterior cingulate compared to somatosensory cortex.
In the hippocampus of GCLM (−/−) mice, PV-IR interneurons also showed a high vulnerability to oxidative stress. The number of PV-IR interneurons was normal in 20-day-old GCLM (−/−) mice, but was strongly reduced in the ventral but not dorsal hippocampus of 4–5-month-old GCLM (−/−) mice (103). The decrease in PV-IR interneurons was particularly selective for the ventral CA3 and dentate gyrus, and this impairment emerged after weaning age as oxidative stress increased or cumulated selectively in the ventral hippocampus (103).
These observations confirm that PV-IR interneurons are particularly sensitive to a GSH deficit but their vulnerability depends on brain regions and correlates with the level of oxidative stress. This also supports the notion that PV-IR fast-spiking interneurons are highly vulnerable to oxidative stress (106). Thus, signs of DNA oxidation and more severe impairment of PV-IR interneurons were observed in the anterior cingulate cortex and the ventral hippocampus, but not in the somatosensory cortex and the dorsal hippocampus (103; Cabungcal et al., unpublished). Interestingly, the anterior cingulate cortex and the anterior hippocampus (analogous to the ventral hippocampus of rodents) are also two brain regions that are known to be affected in schizophrenia (107, 108).
Myelin . Preliminary results suggest less myelination in the anterior cingulate cortex of GCLM (−/−) mice, as revealed by weaker MBP immunolabeling intensity and thinner MBP-IR profiles (105). Further investigations are currently under way to identify anomalies associated with myelin and oligodendrocytes. Indeed, redox state modulates proliferation and differentiation of oligodendrocyte precursors (109). Thus, mice with a chronic deficit in GSH show impaired fast-spiking PV-IR interneurons and altered myelination, both of which occur in schizophrenia (69, 110).
Reduction of γ-Oscillations
Fast-spiking PV-IR interneurons control the output of principal neurons and are necessary for the generation of γ neuronal synchrony that facilitates information processing and transfer within and between brain regions during cognitive tasks (111). Such γ-oscillations are reduced in schizophrenic patients during impaired cognitive tasks (112, 113), but also at resting state (114). This suggests that anomalies in synchronized neuronal activity, driven by PV-IR interneurons, are a core feature of the disorder. Therefore, we examined whether the reduced number of PV-IR interneurons in the ventral hippocampus of GCLM (−/−) mice was also associated with reduced γ-oscillations. We found that β/γ-oscillations induced by kainate in CA3 were significantly reduced in ventral but not dorsal hippocampal slices of GCLM (−/−) mice (103). These results indicate that a chronic deficiency in GSH affects PV-IR interneurons and the generation of β/γ-oscillations in the ventral but not dorsal hippocampus. Such anomaly of synchronized neuronal activity in specific brain regions is therefore expected to affect the behavior accordingly.
Behavioral Phenotype
A chronic GSH deficit did not affect in an unspecific and broad manner all behaviors investigated to date. Overall, the behavioral differences between GCLM (−/−) and (+/+) mice were quite subtle. Using an array of paradigms that require an intact hippocampus, we found a selective alteration in behaviors that rely on the ventral rather than the dorsal hippocampus. GCLM (−/−) mice showed intact spatial reference learning and memory in a water maze and intact performance in a rewarded alteration task (103), indicating normal spatial learning and memory capacity and functionally intact dorsal hippocampus (115). By contrast, tasks that are modulated by the ventral hippocampus (115) were altered in GCLM (−/−) mice. Thus, GCLM (−/−) mice showed a lack of behavioral inhibition in stressful conditions. Compared to wild-type mice, GCLM (−/−) mice entered significantly more often the open arms of an elevated plus maze, performed significantly more transitions between the two compartments of a light/dark box, and spent more time in the anxiogenic light compartment (103). GCLM (−/−) mice also displayed reduced acquisition of delay fear conditioning and expression of contextual and auditory-cued fear, again suggesting a functional anomaly of the ventral but not dorsal hippocampus (103). The ventral hippocampus is indeed implicated in both contextual and auditory-cued delay fear conditioning, while the dorsal hippocampus is involved in trace fear conditioning and contextual delay fear conditioning, but not auditory-cued delay fear conditioning (116–118). Moreover, GCLM (−/−) mice showed novelty-induced hyperactivity and strong exploratory drive toward novel objects. Despite this increased object exploration, however, GCLM (−/−) mice displayed impaired object recognition memory. Interestingly, adult rodents with a BSO-induced GSH deficit during postnatal development or an acute GSH deficit at adulthood also had impaired object recognition abilities (51, 64, 65) but intact spatial reference learning and memory in a water maze ((86); Schenk et al., unpublished).
Further characterization of the behavioral phenotype of GCLM (−/−) mice focusing on startle responses and PPI, responses to stress, and specific cognitive tasks requiring functional PFC is under current investigation.
3.2 Other Genetic Models of GSH Deficiency
As genetic and functional evidence of an implication of the GCLC in schizophrenia exists, it would be of interest to study animals with a genetic defect on GCLC. Knockout of GCLC gene is however lethal at early embryonic stage (5, 98). GCLC (+/−) mice, which are viable and display a moderate GSH deficiency (in the liver: 20 and 50% decrease of GSH and GCL activity, respectively (98); but no published data on brain GSH levels), could be another model worth investigating.
Similarly, mice knockout for the excitatory amino acid carrier-1 (EAAC1) is also an interesting model of GSH deficiency. Observations of altered expression of EAAC1 (=EAAT3) are reported in post-mortem brains of schizophrenic patients (119, 120). EAAC1, which transports glutamate and cysteine (the limiting precursor for GSH synthesis), is specifically expressed in neurons. EAAC1 (−/−) mice have a neuronal deficiency in GSH, leading to increased susceptibility of neurons to oxidant injury (121). Cerebral cortex and hippocampus of these mice show particularly elevated oxidative stress. EAAC1 (−/−) mice have also ventricular enlargement and brain atrophy, which progress with aging (121).
The Na+-independent glutamate–cysteine exchanger (xCT) plays a crucial role in astrocytes and developing neurons to provide them with cysteine, the limiting precursor of GSH. Interestingly, xCT has been implicated in cocaine relapse (122). Sut/sut mice (xCT loss-of-function mutants) could also represent a valuable model. Interestingly, these mice develop brain atrophy by early adulthood, exhibiting ventricular enlargement, thinning of the cortex, and shrinkage of the striatum (123), as observed in schizophrenia.
3.3 Models of GSH Deficit to Investigate Comorbidity with Somatic Disorders
Alterations of the GSH system in schizophrenia have been found in not only brains but also blood and skin fibroblasts (15, 25, 27, 30, 96, 124). Therefore, a genetically compromised GSH system can affect all tissues and could represent a vulnerability factor for not only psychiatric diseases but also several somatic disorders. Genetic polymorphisms of GCL genes have been associated with various pathologies, including cardiovascular diseases (125–127) which are more prevalent in schizophrenia (128, 129).
Compared to the general population, patients with schizophrenia also suffer more often from type 2 diabetes mellitus and have higher frequency of impaired glucose tolerance (130). Thus, patients with schizophrenia and relatives have higher levels of baseline plasma levels of insulin compared to control subjects. But 2 h after administration of oral glucose, levels of both glucose and insulin are higher in patients and relatives (130), suggesting a blunted response to insulin. These observations suggest either shared environmental or genetic predisposition in schizophrenia and in the impaired glucose tolerance. Interestingly, a dysregulation of the GSH system could be one candidate as a common vulnerability factor. Indeed, elevated oxidative stress and low GSH levels were reported in patients suffering from diabetes (131, 132). Hyperglycemia and insulin deficiency can lead to decreased GSH levels via reduced expression of GCLC (see (7)). Alteration in the mutual interactions between the GSH system and the glucose metabolism might be central to the observed comorbidity between schizophrenia, and more generally to the metabolic syndrome in schizophrenia (133–136). Further investigations on the role of the GSH system in the comorbidity of schizophrenia with various somatic disorders are needed. In this context, GCLM (−/−) mice and other genetic models of GSH deficit could be used to address these aspects.
4 Summary
Data collected (Table 7.1) using various animal models of GSH deficiency have revealed the following:




A GSH deficit affects the brain in a region-specific manner. The PFC (in particular the anterior cingulate cortex) and the ventral hippocampus are especially vulnerable, displaying elevated oxidative stress and impairment of fast-spiking PV-IR interneurons.
A GSH deficit renders the brain more vulnerable to high levels of catecholamine release, in particular dopamine. The most vulnerable brain regions to a GSH deficit appear to be regions that are densely innervated by catecholaminergic neurons.
A GSH deficit causes complex changes in dopaminergic transmission as evidenced by enhanced D2R-dependent signaling and enhanced amphetamine-induced dopamine release in the nucleus accumbens.
A GSH deficit causes hypofunction of NMDA receptors.
A GSH deficit during development disrupts cortical circuit in the anterior cingulate cortex, leading to permanent morphological anomalies (i.e., reduced number of PV-IR interneurons and altered myelination) and long-term behavioral impairment in object recognition and multi-sensory processing, but not in spatial memory and learning in water maze.
A chronic GSH deficit also causes a loss of PV-IR interneurons in the adult ventral hippocampus as oxidative stress increases selectively in this region after weaning age. Such impairment of PV-IR interneurons in the ventral hippocampus is accompanied by reduced β/γ-oscillations.
A chronic GSH deficit leads to impaired object recognition, inadequate response to emotion and stress-related situations with a lack of behavioral inhibition, and reduced responses to fear. Under acute GSH deficiency, adult animals display altered responses to psychostimulants and impairment in object recognition. Finally, a transitory GSH deficiency during postnatal development is sufficient to cause long-term behavioral effects, affecting mostly object recognition and multi-sensory processing. Overall, all these types of GSH deficit lead to impaired object recognition but tend to leave spatial abilities intact when tested in a water maze.
Table 7.1
Summary of the main effects of different induced GSH deficits on general health parameters, brain morphology, physiology and neurotransmission, and behavior in rodents. ACC, anterior cingulate cortex; SSC, somatosensory cortex; HP, hippocampus
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
