Chapter 30 Glucocorticoids and Mineralocorticoids
Physiology: Control of Endogenous Glucocorticoid Secretion
The adrenal cortex comprises three zones, each of which synthesizes steroidal hormones. From superficial to deep, these include the zonas glomerulosa, the source of mineralocorticoids (predominantly aldosterone but also corticosterone); reticularis, a source of (weak) androgens; and fasciculata, the source of GLCs (cortisol and cortisone). As with many hormones, the secretion of corticosteroids reflects a balance between positive and negative feedback pathways. Corticotropin-releasing hormone (previously corticotropin release factor, CRH) is secreted by the hypothalamus and travels through the hypophyseal portal system to the adenohypophysis, where it stimulates the synthesis and secretion of adrenocorticotropin (ACTH) from the basophilic cells of the adenohypophysis (Figure 30-1). In addition to its role in promoting ACTH secretion, CRF appears to be involved in the autonomic, immunologic, and behavioral response to stress independent of the hypothalamopituitary axis. Administration of CRF to dogs results in a decrease in gastric acid secretion by activation of the sympathetic nervous system, an immediate decrease in mean arterial blood pressure accompanied by reflex tachycardia, and a marked increase in plasma vasopressin concentrations.1 These actions of CRF are independent of GLCs.
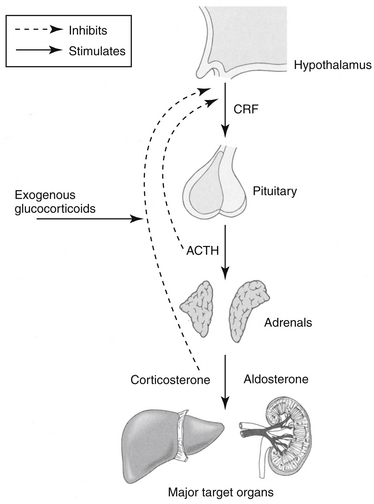
In addition to CRF, ACTH also is stimulated by arginine vasopressor (AVP), which is a weak stimulator of ACTH but a strong stimulator of CRH and catecholamines; angiotensin II; serotonin; and vasoactive intestinal peptide. Other stimulators include the inflammatory cytokines interleukin (IL) -1, -2, and -6 and tumor necrosis factor-α(TNF-α).2 Most stimulators of ACTH also stimulate CRH.2 The primary short-term effects of ACTH are stimulation of the adrenal cortex synthesis and secretion of cortisol, corticosterone, aldosterone (the effect of ACTH on mineralocorticoid secretion is minimal), and weak androgenic substances. Long-term effects of ACTH increase the production of enzymes and cofactors necessary for cortisol production and cause an increase in adrenal receptors for low-density lipoprotein cholesterol.2
Cortisol and corticosterone concentrations in plasma subsequently influence CRF and ACTH secretion such that increased concentrations inhibit release of CRF and ACTH and reduced concentrations stimulate release of CRF and ACTH. Exogenous factors, such as trauma, heat, stress, surgery, and neural impulses, also mediate CRF and ACTH secretion. Exogenous corticosteroid administration can also suppress CRF and ACTH release. The degree of suppression depends on the particular drug used. For example, the synthetic drug dexamethasone is 50 to 100 times more potent in suppressing ACTH secretion than is the endogenous compound, cortisol.3 The diurnal variation in GLC secretion that occurs in humans has not been well documented in dogs or cats. Nonetheless, morning dosing (in dogs) is among the strategies used to minimize the risk of adrenocortical suppression that might result from exogenous glucocorticoid therapy. These approaches thus include: 1. Determining a minimum effective dose; 2. Alternate day dosing; 3. Use of a “short-acting” glucocorticoids whose effects are 24 hrs or shorter in duration; 4. Dosing in the morning (and potentially dosing at nights for cats); and 5. Tapering the dose over days to weeks as therapy is discontinued, thus facilitating readaptation of the adrenal gland to secretion.
Mechanism of Action
The myriad physiologic effects of GLCs result from interaction of the drugs with the glucocorticoid receptor (GR), one member of a nuclear hormone receptor superfamily that also includes receptors for thyroid hormone, mineralocorticoid, estrogen, and progesterone.4 Other activities may also reflect nonreceptor mechanisms.
Mineralocorticoid and Glucocorticoid Receptors
Two primary types of corticosteroid receptors exist. Type I, or mineralocorticoid receptors (MRs), bind both endogenous GLCs and aldosterone. Type II, or GRs, bind endogenous and exogenous GLCs but have a poor affinity for mineralocorticoids. The GRs and MRs are sufficiently similar that drugs may bind to both, causing similar responses. In human medicine, the cause and effect relationship between hypertension, heart failure, and MRs has led to a reassessment of the relationship between GLC and mineralocorticoid as well as androgen and progesterone receptors.5 The distinction among the receptors, including the sequence of their evolution, becomes important when assessing the selectivity or lack thereof of therapeutic agents on each receptor type. In human medicine this is particularly germane to the impact of GLCs on the MRs and its implication in patients with cardiovascular disease.
Every cell appears to have GRs, although the liver is the primary target. The type and concentration of GRs varies between species and tissue. Type II receptors are more ubiquitous in the brain. The degree to which GLCs bind to each receptor type varies with the circulating concentration. At basal levels type I receptors are preferred, but as cortisol concentrations increase (e.g., during stress), type II receptors increasingly are activated.6
Within a given tissue, GR numbers appear to fluctuate with changing cell cycles and age and in response to a variety of endogenous or exogenous compounds. More than 15 endogenous regulators have been identified for GLC receptors.7 Response to GLCs reflects receptor density or GLC concentration, depending on the tissue. GR density is autoregulated; increased receptor density associated with hypoadrenocorticism can be reversed with GLC replacement.8 Likewise, chronic GLC therapy will result in downregulation of receptor density. However, autoregulation is tissue specific, and in some tissues the concentration of GLC is a more important determinant of response than is the number – or even type – of receptors. For example, as noted, the MR acts similarly to the GR and is sufficiently similar in structure that responses to substrates are also likely to be similar.8,9 Any steroid with a ketone group at position 11 (Figure 30-2) will interact positively with either the MR or GR.2 However, GLCs generally are present in concentrations that exceed those of mineralocorticoids by 1000-fold or more,2 which suggests the potential for GLC response to predominate in any tissue exhibiting either GRs or MRs. The ability of receptors to respond to mineralocorticoids despite differences in concentration reflects a combination of conditions: (1) binding of glucorticoids by globulins, which effectively sequesters the hormone, precluding interaction with GRs or MRs; (2) activation of discrete sets of target genes, perhaps reflecting a difference in repression of transcriptional induction (e.g., aldosterone activates the expression of several genes, with that of Na+, K+-ATPase in the basolateral membranes of tubular cells being the best characterized); (3) restrictive expression of MRs only, with the principle sites occurring in the renal cortical distal tubules and collecting ducts, colon, salivary and sweat glands, and hippocampus8; (4) differing rates of GLC inactivation, which allows mineralocorticoids to predominate and stimulate response. For example, tissues differ in their expression of 11 β-hydroxysteroid dehydrogenase type 2 (11 β-HSD2), the enzyme that inactivates cortisol to corticosterone. Tissues with high concentrations of 11 beta-HSD2are those that typically respond to mineralocorticoids—kidney, colon, salivary gland—allowing mineralocorticoid effects to predominate.8,10 In the presence of 11 β-HSD2, GLC expression occurs only if present in concentrations several magnitudes higher than mineralocorticoids.
Certain disease conditions may reflect aberrations in methods allowing differential substrate response such as an absence of 11 β-HSD2 such that tissues respond to the inappropriate steroid.9 The importance of renal metabolism of cortisol is implied in the impact renal failure has on prolongation of plasma cortisol half-life in human patients; half-life increased 30% in one study.10 Impaired 11 β-HSD2 accompanying renal disease contributes to the pathophysiology of salt and water retention and hypertension in renal failure and therefore could contribute to selected consequences of renal disease (hypertension, sodium retention, hyperkalemia, and decreased glomerular filtration rate). Inhibition of 11β HSD2by selected compounds (licorice, selected angiotensin-converting enzyme inhibitors [ramipril]) contributes to pharmacologic or adverse events as well as drug interactions. Ramipril causes decreased ACTH-induced hypertension in the rat.10 Inhibition allows GLCs to excessively stimulate MRs such that hyperaldosteronism emerges. Such interactions also can be favorable:
Nuclear and Non-nuclear Mechanisms of Action of Corticosteroids
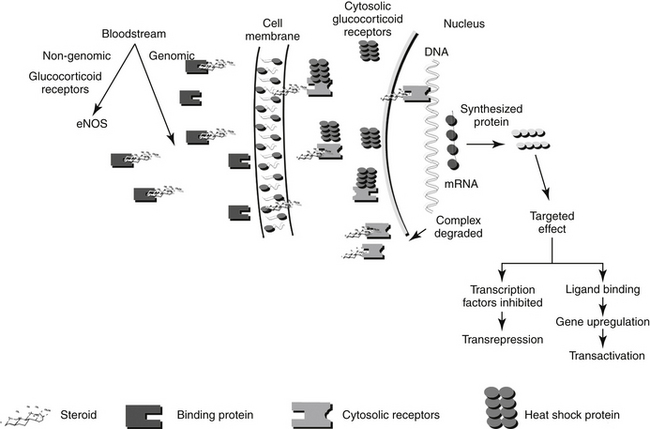
The molecular description of the GR as it relates to its mechanism of interaction has been described.11–14 The GR consists of at least two isoforms.11 The α isoform binds to GLCs, DNA, and transcription factors and thus modulates transcription activity. The α isoform may also act through nongenomic mechanisms. In contrast, although the β isoform binds to DNA, it does not bind other ligands, fails to activate transcription, and appears to be able to interfere with the activities of α isoforms.12 Corticosteroid receptors are located in the cytoplasm of the target cell complexed with heat shock proteins (Hsp 70 and Hsp 90) that act as chaperones, proteins which in non-functional activities (e.g., folding, assembly) of macromolecules, and an immunophilin, an intracellular protein that binds other immunosuppressive compounds.9,11 The complexed receptor is inactive until bound to a steroid ligand.9 Steroids enter the cells by passive diffusion, although a rate-limited active transport mechanism may also exist (Figure 30-2) Once in the cell, the GLC binds to the receptor, causing the heat shock protein and other molecules to dissociate. Intracellular GLCs impart their effects in one of three ways: The steroid–receptor complex moves into the nucleus and (1) binds to specific DNA sequences causing the regulation of genes (for example, lipocortin or genes responsible for gluconeogenesis) (referred to as a “cis” or element, resulting in transactivation), (2) interacts with other transcription factors (e.g., AP-1, NFκB), generally at lower concentrations, that prevent the transcription of targeted genes (e.g., TNFα, IL-1B, IL-4 and 5; referred to as a “trans element” resulting in transrepression); or (3) through non-genomic mechanisms involving membrane-associated GR and secondary messenger systems.2,12a Non-genomic responses are rapid in onset, and includes vascular responses (eNOS activation), lymphocytolysis, and possibly, induction of inflammatory prostaglandins. “Permissive” effects of GLC may reflect non-genomic responses.
Because the effect of older, current and future GLC reflects the molecular relationship between drug and cellular macromolecules, a brief review or molecular interactions is warranted. Genomic effects of GLCs are initiated by translocation of the activated receptor–GLC complex to the nucleus (Figure 30-2). In the nucleus the complex binds to regulatory proteins of target genes. The short DNA sequences recognized by the activated GR are referred to as glucocorticoid responsive elements (GREs). It is through these GREs that specificity of GLC-modulated gene transcription is controlled.4 Transcription of the gene and subsequent formation of the targeted protein is either induced (transactivated) or inhibited (transrepressed). The receptor and GLC are eventually metabolized (the exact location or timing is not documented). The cellular half-life of the activated complex is about 10 hours.15 It is not known whether the rate of metabolism of the GLC–receptor complex is dependent on the specific GLC involved. Some of the proteins that are regulated by GLCs are listed in Table 30-1. The proteins encoded by these genes are responsible for some of the physiologic (pharmacologic) effects of GLCs.
Table 30-1 Immunomodulatory Actions of Glucocorticoids
| Actions | Mechanism |
|---|---|
| Interference with cytokine production | 1 |
| Interleukin-1 | |
| Interleukin-2 | |
| Interleukin-6 | |
| Interleukin-8 | |
| Tumor necrosis factor-alpha | |
| Interferon-gamma | |
| Downregulation of cell adhesion molecules | 1 |
| Intercellular Adhesion Molecule 1 (ICAM-1) | |
| E-selectin | |
| Inhibition of chemokine synthesis | |
| Interference with leukocyte function enzymes | 1 |
| Inducible nitric oxide synthase | |
| Granzyme B | |
| Upregulation of cytokine receptors | |
| Interleukin-1 | |
| Interleukin-6 | |
| Induction of lymphocyte apotosis | |
| CD4+CD8+ | |
| Inhibition of cyclooxygenase-2 expression | |
| Inhibition of wound healing | 2 |
| Epithelial cells | |
| Fibroblasts | |
| Induction of acute phase proteins (permissive effect) | 3 |
| Serum amyloid A | |
| Serum amyloid P | |
| AP-1 | |
| NFk-B | |
| Stat proteins |
1 = protein-protein interaction with transcription factors
Once activated, the activated ligand receptor influences gene expression by binding to specific GREs in the promoter regions of GLC-regulated genes. However, in addition to this classical transactivation mechanism, also referred to as positive GRE (pGRE), genes may also be negatively targeted, or transrepressed, through negative GRE (nGRE). The GR generally has a lower affinity for nGRE than pGRE. Transrepression involves protein–protein coupling to other transcription factors (e.g., nuclear factor kappa-B [NFκB], activator protein-1 [AP-1], STAT-5 (signal transducer and activator of transcription) or nuclear factor of activated T-cells [NFAT]) such that their activity is modulated. The importance of these different GR interactions results in differential physiologic (and pharmacologic) effects and ultimately provides a basis for directed drug therapy. The largely undesirable metabolic effects of long-term GLC therapy appear to be mediated by pGRE interaction (i.e., transactivation gene expression). In contrast, the desirable antiinflammatory effects appear to reflect modulation of transcription factors (i.e., transrepression through interaction with nGREs).16,17 Indeed, nGREs contribute to the regulation of the hypothalamic–pituitary–adrenal axis (targeting pro-opiomelanocortin [POMC] and CRH), bone metabolism (through osteocalcin), skin function, inflammation (IL-1β), angiogenesis (proliferin), and lactation (prolactin).17 Trans (protein-coupling) effects, and repression in particular, appear to occur at concentrations lower than that necessary for cis (transactivation) effects,18 providing another mechanism of directed therapy. “Designer” GLC might be developed such that potency for protein coupling (trans) repression is greater than DNA interaction, leading to cis or trans activation. Dexamethasone and prednisolone are examples of a “symmetric” GLC, characterized by equal binding affinity for both actions. Interestingly, medroxyprogesterone acetate exhibits a preponderance of transrepression activity rather than transactivation.16 Newer GLCs are likely to have a preponderance of activity for either activation or repression. The recognition of this dual mechanism of GLC action will also facilitate the development of GR antagonists, which might be useful for the treatment of Cushing’s disease.16 Another term that has emerged is “Dissociated GLCs” which target GLC-dependent transactivation or alternatively act as inhibitors of NFκB-dependent transcription, thus retaining their in vivo antiinflammatory activity but avoiding their undesirable metabolic effects.19
Differences in GR affinity for nGREs versus pGREs may also offer a mechanism for differential targeted drug response based on dose (or concentration). Treatment with low, rather than high, doses of GLCs appears to minimize undesirable side effects.20 Using a retrospective analysis of published literature (meta analysis precluded by method variability) coupled with a prospective study, researchers found that most adverse effects to GLCs in humans were associated with high, rather than low, doses (7.5 mg prednisolone equivalent in humans [approximately 0.1 mg/kg]) of GLC (see the section on adverse events).
Glucocorticoid Resistance
Failure to respond to steroid therapy is observed in human patients being treated for a variety of diseases. Poor compliance and low bioavailability must be ruled out before other factors are considered. The documented phenomenon of GLC resistance (both familial and iatrogenic or drug induced forms have been reported in humans) may reflect, among other causes, reduced receptor numbers or affinity for GLC.21 Cellular response to GLCs has been directly correlated with receptor numbers. Reversible downregulation of receptor numbers and a subsequent decrease in biological effect are documented sequelae of GLC treatment.7 Such effects have been demonstrated on lymphocyte receptors.22 GLC failure in patients receiving long-term GLC therapy for treatment of host-versus-graft rejection has been attributed to downregulation of cytoplasmic GRs in T lymphocytes. GLC doses subsequently must be increased.23 Pulsing of high doses of GLCs may overcome this relative resistance (see the section on therapeutic use).24 The relative balance of the α and β isoforms of GR appear to influence cell sensitivity to GLCs; higher concentrations of β contribute to GLC resistance.12 Treatment of inflammatory bowel disease offers an example of potential mechanisms of GLC resistance. Some human patients with severe disease are nonresponsive to even high doses of GLCs. Lack of response has been associated with poor antiproliferative effects on blood T lymphocytes, in contrast to near-complete inhibition of responders. A similar relationship has been demonstrated for other chronic allergy-based diseases, such as asthma or rheumatoid arthritis, and renal allograft rejection. A relatively fast in vitro T lymphocyte proliferation assay has been useful in identifying nonresponders before treatment.25 Several molecular mechanisms have been proposed to account for poor control of T cell proliferation by GLCs. These include overexpression of the multidrug resistance gene (MDR1 polymorphism), resulting in increased P-glycoprotein–mediated efflux of cytoplasmic (see relevant discussion in the section on neoplasia). This mechanism might be reversible by co-administration of cyclosporine, an inhibitor of P-glycoprotein. Other mechanisms include impaired GLC signaling by the GR and increased activation by epithelial cells of proinflammatory mediators that directly inhibit GR transcription. This latter mechanism (postreceptor failure) is supported by the presence of clinical signs consistent with Cushing’s disease in humans with poor inflammatory control.25 Differences in activation patterns of NFκB have been associated with GLC resistance in human patients with chronic inflammatory bowel disease.19 Ultimately, differences in response at the molecular level may also be shown .
Physiologic Effects
Physiologic effects of GLCs required for the “normal” day-to-day function of the animal can be easily appreciated when GLCs are absent, as in the case of adrenocortical insufficiency (hypoadrenocorticism or Addison’s disease), or present in excessive concentrations, as in hyperadrenocorticism. The primary role of aldosterone, the major mineralocorticoid, is regulation of sodium homeostasis. Interestingly, the effects of GLCs and mineralocorticoids can be directly antagonistic at some sites. For example, in some tissues aldosterone stimulates and GLCs inhibit sodium resorption.8 Likewise, cortisone can have different and opposing effects, depending on whether it interacts with MRs or GRs. Cortisone inhibits microglial proliferation through GRs but stimulates proliferation through MRs.26 Dexamethasone also inhibits microglial cell proliferation at concentrations lower than those for cortisone.26
Effects on Intermediary Metabolism: Carbohydrates, Proteins, and Lipids
The natural function of GLCs is to protect glucose-dependent cerebral functions by stimulating the formation of glucose by the liver, decreasing its peripheral utilization and promoting its storage as glycogen.27 Teleologically, these effects protect glucose-dependent tissues, the brain and heart, from starvation.9 The hyperglycemic effect of GLCs, as seen in the stressed patient, is due to an increase in gluconeogenesis and insulin antagonism. Gluconeogenesis is the result of an increase in precursors necessary for gluconeogenesis as well as induction of hepatic enzymes that catalyze reactions of glucose synthesis. Increased breakdown of proteins, particularly skeletal muscle and collagen, provides gluconeogenic precursors (e.g., amino acids and glycerol). This effect is exhibited clinically as muscle wasting, delayed wound healing, and thinning of the skin. The anti-insulin effect of GLCs is a result of decreased peripheral tissue utilization of glucose and reduced affinity of cellular receptors for insulin. Utilization appears to be decreased by translocation of insulin receptors from the cell membrane to an intracellular location inaccessible by insulin.9
Metabolism of lipids is also affected by GLCs. Specifically, GLCs promote lipolysis, generating the free fatty acids that, along with amino acids, serve as substrates for hepatic glycogen synthesis and inhibit long-chain fatty acid synthesis. Effects of GLCs on lipid metabolism reflect, in part, a permissive effect of the steroids on other agents, including growth hormone and β-adrenergic receptors. One sequela of these effects is redistribution of body fat (such as is typified by Cushing’s disease). Differences in adipocyte sensitivity to insulin and the facilitating effects of GLCs may explain the redistribution phenomenon.9
Water and Electrolyte Balance
Cortisol appears to be essential for the maintenance of renal blood flow and glomerular filtration rate. Volume repletion alone is not sufficient to return renal function to normal in states of hypoaldosteronism. GLCs also generally increase the filtration fraction; impact on renal vascular resistance is not clear, varying with the method studied.10 Aldosterone is the most potent natural corticosteroid that affects fluid and electrolyte balance. Mineralocorticoids act to enhance sodium reabsorption in exchange for potassium (from the distal renal tubules and collecting ducts) or hydrogen (the intercalated cells), resulting in a positive sodium balance, expansion of extracellular fluid volume, and an increase in glomerular filtration rate. Sodium reabsorption is enhanced by increasing the number of open Na+ and K+ pores; Na+, K+-ATPase activity at the basolateral membrane also is increased, causing sodium to be returned to the circulation. The ratio of exchange of these mono-charge cations may be greater than than 11:1.
Other reported renal actions of GLC include downregulation of sodium–phosphate co-transport and sulfate co-transport, upregulation on sodium–bicarbonate co-transport, modulation of the effects of sodium on amino acid transport, and other effects on acid–base balance.10 GLCs also influence several aspects of calcium movement. In the renal tubular cells, calcium excretion is increased, and in the small intestine its absorption is impaired. GLCs also increase parathormone secretion, which in turn increases osteoclast-mediated bone resorption. The net effect of GLCs on calcium homeostasis is a decrease in total body calcium stores.
Hemolymphatic System
GLCs tend to increase the red blood cell content of the blood by retarding erythrophagocytosis. Lymphopenia, eosinopenia, and monocytopenia caused by cellular redistribution and neutrophilia caused by increased release from bone marrow, demargination, and a reduction of their removal from the circulation are all associated with GLC administration.9 This blood cell profile represents the “stress leukogram” seen clinically in patients with increased concentrations of endogenous GLCs. The acute effects of GLCs on circulating lymphocytes are due to sequestration from the blood rather than lymphocytolysis, although cells of lymphocytic malignancies are destroyed by GLCs. In addition to reducing the number of circulating lymphocytes, GLCs also alter the responses of lymphocytes to mitogens and antigens. T lymphocytes are inhibited to a greater degree than B lymphocytes (see following discussion of immunomodulation). GLCs also induce lymphocyte apoptosis.28 GLCs also have direct inhibitory effects on eosinophils, including eosinophil migration.29 GLCs directly induce eosinophil and lymphocyte apoptosis.30
Antiinflammatory and Immunosuppressive Effects
GLCs are most frequently used in clinical medicine for their antiinflammatory and immunosuppressive actions (see Table 30-1).31 Because the antiinflammatory and immunosuppressive effects of GLCs reflect specific actions on white blood cells, these effects are inextricably linked. They generally occur only when the amounts of steroid are present in concentrations greater than those found in the normal physiologic state (i.e., pharmacologic concentrations). The effects of GLCs on leukocyte numbers were discussed earlier. GLCs also profoundly affect white blood cell function. Ultimately, both the humoral and cell-mediated arms of the immune response are affected. The antiinflammatory effects of GLCs reflect all three mechanisms of GLC action (genomic transcription and transactivation; nongenomic membrane receptor signaling).12
GLCs upregulate or downregulate as many as 2000 genes involved in the regulation of the immune response (Table 30-4).2 GLCs inhibit early and late phases of the inflammation. Responses that are inhibited include edema formation, fibrin deposition, leukocyte migration, phagocytic activity, collagen deposition, and capillary and fibroblast proliferation. Many of these processes involve lymphokines and other soluble mediators of inflammation, and it is through these mediators that GLCs exert their antiinflammatory actions. GLCs induce annexin I (also known as lipocortin), which inhibits phospholipase 2, thus blocking the release of arachidonic acid and its subsequent conversion to eicosanoids (i.e., prostaglandins, thromboxanes, prostacyclins, and leukotrienes). (Figure 30-3). GLCs also preferentially inhibit transcription of cyclooxygenase-2 (COX-2), the inducible form of cyclooxygenase.12,32,33 Interestingly, recent evidence indicates that under some circumstances, GLCs may actually induce COX-2.34 The net effect of specificity to inhibit COX-2 may be inhibition of inflammatory prostaglandins without negatively affecting the protective effects of prostaglandins in other body systems (e.g., gastrointestinal, renal, hemostasis). GLCs induce the protein MAPK phosphatase 1, which through various actions inactivates a number of proteins important in the signaling of cytokines.12 Through physical interaction, transcription of NFκB is inhibited by GLCs. Many of the antiinflammatory effects of GLCs occur rapidly, apparently independently of changes in gene expression (nongenomic). Examples include activation of endothelial nitric oxide synthetase,12 offering an alternative mechanism of action of a new class of more selective antiinflammatory drugs.35
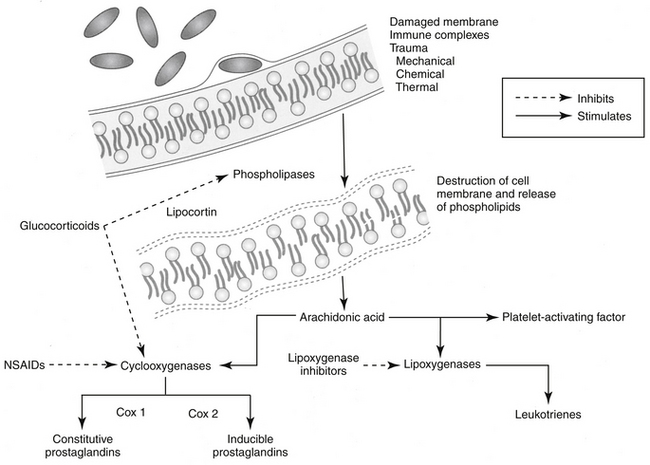
Among the sequelae of these mechanisms, GLCs inhibit release of TNFα and IL-2 from activated macrophages. TNFα induces cytotoxicity and can enhance neutrophil and eosinophil function.27 The release of platelet-activating factor from leukocytes and mast cells is inhibited by GLCs. Platelet-activating factor induces vasodilation, platelet and leukocyte aggregation, smooth muscle contraction (especially in the bronchi), and increased vascular permeability.36
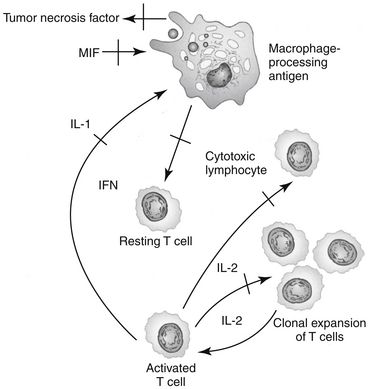
The immunosuppressive actions of GLCs, like their antiinflammatory actions, involve disruption of intercellular communication of leukocytes through interference with lymphokine production, biological action, or both. GLCs inhibit the synthesis and release of IL-1 by macrophages, thereby suppressing the activation of T cells. GLCs also inhibit IL-2 synthesis by activated T cells. IL-2 plays a critical role in amplification of cell-mediated immunity. GLCs block the effects of the migration inhibitory factor-γ (causing macrophages to migrate away from the affected area) and interferon-γ (IFN-γ) on macrophages.27 IFN-γ, which is released from activated T cells, plays an important role in facilitating antigen processing by macrophages. Additionally, GLCs suppress the bactericidal and fungicidal actions of macrophages. GLCs also alter synthesis of and biologic response to collagenase, lipase, and plasminogen activator. The antiinflammatory effects of GLCs also may reflect inhibition of the inducible form of nitric oxide synthase (iNOS).37 Synovial macrophage nitric oxide production and iNOS synthesis are inhibited by dexamethasone. The inhibitory effect appears to be mediated by lipocortin.The immunosuppressive actions of GLCs are more pronounced on the cellular arm than the humoral arm of the immune system (Figure 30-4). GLCs have minimal effects on plasma immunoglobulin concentrations but can modulate immunoglobulin function. For example, opsonization of bacteria is inhibited. Therapeutic doses of GLCs do not significantly decrease an animal’s antibody response to antigenic challenge (e.g., vaccinations). The immunosuppressive effects of GLCs may also reflect actions on the hypothalamic–pituitary–adrenal axis. Multiple cytokines appear to regulate this axis. Specifically, IL-1 appears to stimulate the release of CRH and directly increase the release of ACTH, and it may cause the adrenals to release GLCs. These interactions appear to be important to modulation of stress and thus maintenance of homeostasis.9
Cardiovascular System
Corticosteroids have two major effects on the cardiovascular system. Mineralocorticoids and, to a lesser extent, GLCs affect the maintenance of extracellular fluid volume, as described previously. Interestingly, mineralocorticoids also appear to have direct actions on cardiovascular tissues; increased cardiac fibrosis has been induced experimentally in rats by administration of excessive mineralocorticoids, suggesting an indication for spironolactone as a diuretic in congestive heart failure associated with myocardial disease.9 In addition, corticosteroids (predominantly GLCs) enhance vascular reactivity to other vasoactive substances (e.g., norepinephrine, angiotensin II).9 Mechanisms appear to include increased receptor numbers in the vascular wall or other tissues. Other proposed mechanisms of GLC-induced hypertension include reduced activity of depressor systems (e.g., kallikrein-kinin, prostaglandins, and nitric oxide) and increased responsiveness to angiotensin II and norepinephrine.38 In humans, Cushing’s syndrome or prolonged administration of synthetic GLCs is associated with hypertension characterized by sodium chloride retention and volume expansion. However, hypertension may be independent of these effects; rather, downregulation of nitric oxide synthetase may be a more plausible explanation.39 In contrast, in patients with insufficient concentrations of GLCs, negative sequelae include increased capillary permeability, decreased cardiac output, and inadequate vasomotor response of the smaller blood vessels to catecholamines. In humans, glucocorticoids are associated with worsening of cardiovascular disease. Hypertension appears to reflect increased glucocorticoid-mediated peripheral vascular resistance rather than aldosterone–mediated rather than sodium retention. The latter mechanism, along with myocardial remodeling, has been associated with worsening congestive heart failure.39b
Bone and Cartilage
GLCs antagonize the effects of vitamin D3, accelerate bone resorption, and decrease bone formation (through direct action on osteoblasts), resulting in osteoporosis. This phenomenon is well documented in human patients after chronic GLC therapy, but to the authors’ knowledge it has not been observed in animals.40 At physiologic doses, glucocorticoids stimulate collagen. At supraphysiologic doses GLCs inhibit collagen synthesis by fibroblasts; depress chondrocyte metabolism; and decrease the proteoglycan content of cartilage, resulting in morphologic changes in articular cartilage.41,42
Skeletal Muscle
The permissive effects of GLCs include their influence on the ability of skeletal muscle to function normally. Too little will result in muscle wasting (generally resulting from hypokalemia). Likewise, and paradoxically, too much also will result in muscle wasting. Increased use of amino acids from muscle proteins is likely to contribute to this effect. Although the exact mechanisms of muscle wasting are unknown, the term steroid myopathy has been coined to reflect this condition in human patients and is used to refer to similar manifestations in small animals with hyperadrenocorticism.9
Central Nervous System
In the central nervous system (CNS), the highest concentration of MRs is in the limbic system, whereas GRs are more ubiquitous in the brain.6 Indirectly, GLCs maintain adequate plasma concentrations of glucose for cerebral functions, maintain cerebral blood flow, and influence electrolyte balance in the CNS. GLCs decrease formation of cerebrospinal fluid, which results in a reduction of intracranial pressure. In human beings GLCs are believed to influence mood (e.g., producing euphoria), behavior, and brain excitability.9 The euphoric effect commonly recognized in dogs also is likely to reflect differences in GRs. Steroids, including GLCs, also appear to regulate neuronal excitation, with differential effects occurring for some drugs. For example, dexamethasone inhibits microglial cell proliferation; cortisone also will inhibit proliferation at higher doses through GRs, but stimulate proliferation through MRs.26 GLCs appear to have a protective effect in the CNS through induction of glutamine synthetase in both the CNS and peripheral nervous system; the enzyme adds an amine to and thus removes glutamate.43 Increased glutamate has been associated with CNS pathology, although the relationship between the two remains controversial.44,45 However, efficacy reflects regulation through transcription, which takes at least 24 hours. Glutamine has several roles in the CNS; in the presence of hyperammonia (e.g., liver disease) increases formation of glutamine in brain cells and the potenetial for cerebral edema.45b The use of glucocorticoids for treatment of cerebral edema might be avoided in situations in which increased glutamine increases the risk of cerebral edema.
Interestingly, GLC concentrations appear to be directly related to the severity of memory impairment in animals, including humans.6 Learning is impaired in animals exposed to prolonged reductions in circulating concentrations of GLCs; conversely, memory also appears to be impaired if GLC concentrations are too high.
Respiratory System
GLCs are reported to have “permissive” effects (increased receptor number or affinity have been proposed) on β2-receptors, promoting bronchodilation.46 Other mechanisms are likely to evolve as non-genomic effects are understood. The effects of GLCs on leukotrienes, platelet-activating factor, and other mediators important in the pathogenesis of respiratory inflammatory diseases were discussed earlier and also are addressed in Chapters 20 and 29.
Alimentary Tract
GLCs decrease the absorption of calcium and iron from the gastrointestinal tract and increase the absorption of fats. Secretion of gastric acid, pepsin, and trypsin are increased by GLCs. Gastric mucosal cell growth and renewal are reduced by GLCs, and mucus production is decreased, resulting in compromise of the protective barrier of the gastric mucosa. Collectively, these effects contribute to increased susceptibility to gastric ulceration. A retrospective study in humans found that 5% developed gastric mucosal lesions while receiving GLCs, particularly patients with rheumatoid arthritis and collagenosis.47 It is not clear whether the effects of GLCs on the gastric mucosa occur as a result of impaired mucosal prostaglandin synthesis, although failure of misoprostol (prostaglandin E) to protect against GLC-induced ulceration indicates that they do not. Indeed, some studies have shown that GLCs provide a gastroprotective effect during stress-induced ulceration.48 Deposition of glycogen in the liver, resulting in hepatomegaly in animals chronically treated with GLCs, is a consequence of increased glycogen synthesis, as described previously. Excessive cortisol levels can induce pancreatic ductule hyperplasia.
Reproduction
GLCs can induce parturition during the latter stages of pregnancy in ruminants and horses, but their effect in dogs and cats has not been determined. GLC administration in pregnant dogs has been associated with cleft palate and abortion. GLCs have been shown to inhibit cell division, DNA synthesis, or both in the developing liver, lung, brain, and thymocytes.
Structure–Activity Relationship
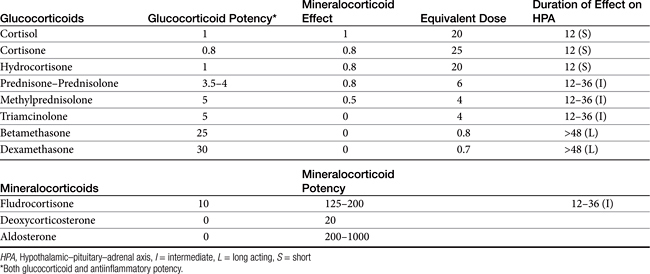
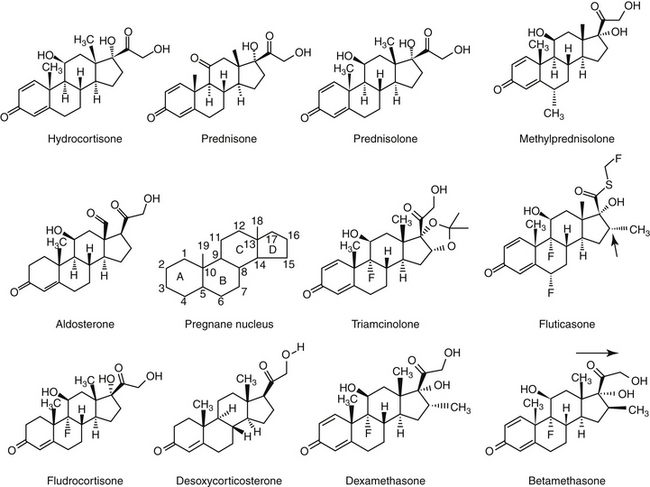
There are close to 50 generic corticosteroid products approved for human use and several approved for use in small animals. These drugs differ primarily in their duration of action, mineralocorticoid activity, and antiinflammatory potency (Table 30-2). As the antiinflammatory potency of a particular agent increases, its biological half-life and duration of action also tend to increase. Antiinflammatory properties also parallel the effects on metabolism, that is, carbohydrate and protein metabolism, but mineralocorticoid effects can be altered independently by changing the molecular structure of the steroid (Figure 30-5). The 4,5 double bond and the 3-ketone are necessary for mineralocorticoid and GLC effects. Synthetic modifications of cortisol increase the antiinflammatory activity, decrease protein binding, and decrease hepatic metabolism, thus prolonging activity. First-generation GLCs formed with the addition of a 1,2 double bond increased the ratio of GLC to mineralocorticoid effects (prednisolone, prednisone, and methylprednisone) and duration of action. The second-generation steroids were fluorinated at the C-9 position, increasing potency. Methylation at the C-16 position eliminates mineralocorticoid activity (dexamethasone, betamethasone, and triamcinolone).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


