Fig. 1.
The Tamoxifen inducible gene switch. (a) Gene inactivation in knockout mice or gene expression in standard transgenic mice is restricted to selected cell types but the timing of gene inactivation or transgene activation cannot be further manipulated. In contrast, in Tamoxifen inducible knockout or transgenic mice, the onset of gene inactivation or transgene expression can be regulated in a selected cell type, e.g. induced in adult mice. (b) The Tamoxifen inducible Cre-ERT2 system. Structure of a transgene construct for cell type-specific expression of the Cre-ERT2 fusion protein. In the Cre-ERT2 fusion gene, the coding region of Cre is fused to the ligand binding domain of the human oestrogen receptor (ERT2, residues 282–595). The ERT2 ligand binding domain harbours three point mutations (***) that render ERT2 unresponsive to the natural ligand estradiol. (c) Principle of the Tamoxifen inducible Cre-ERT2 gene switch. Cre-ERT2 protein is constitutively expressed in a selected cell type but remains inactive by binding to heat shock proteins (HSP90) that interfere with recombinase activity. Cre-ERT2 becomes activated by the ERT2 ligand 4-OH-tamoxifen that leads to the dissociation of HSP90 proteins. Upon HSP90 dissociation, the fusion protein is released from the inactive complex and mediates the excision of a loxP-flanked DNA segment via the active Cre domain. The deletion of a loxP-flanked exon leads to the knockout of the target gene, whereas the deletion of a loxP-flanked stop cassette results in transgene expression.
Mouse genetic technology offers a solution for the timed control of protein expression or gene knockout by the use of a Tamoxifen inducible gene switch that enables the inducible activation of transgene expression and inducible gene knockout. This switch for inducible and cell type-specific gene manipulation is provided by a fusion protein of Cre recombinase with the ERT2 mutant ligand binding domain of human oestrogen receptor (amino acids 282–595, Fig. 1b) (19–21). The CreERT2 fusion protein can be constitutively expressed in mice from a transgene or knock-in allele using a cell type-specific promoter region, but does not exhibit recombinase activity unless a specific activating ligand is supplied. In the absence of this ligand, the ERT2 domain is bound by heat shock proteins that block recombinase activity (Fig. 1c). The ERT2 domain is unresponsive to the natural ligand estradiol due to the change of three amino acid residues. However, recombinase activity of the fusion protein can be induced by the synthetic ligand 4-hydroxy-tamoxifen, which releases the heat shock proteins from the ERT2 domain (Fig. 1c).
Thereby, the CreERT2 protein can be used as an inducible gene switch by the administration of Tamoxifen to mice at a selected time window. The transient activation of the CreERT2 recombinase enables excision of a gene segment from the genome that has been marked for deletion by insertion of two Cre recombinase recognition (loxP) sites (Fig. 1c). The removal of a loxP-flanked exon from an endogenous gene allows the use of the CreERT2 switch for inducible gene inactivation in vivo. Furthermore, the same switch can be used to excise an inhibitory loxP-flanked DNA segment (stop element) from a transgene. The removal of the stop element leads to the expression of the transgene (Fig. 1c).
Thereby the CreERT2 gene switch can be used to generate genetic models of gain-of-function (dominant) disease-associated alleles by the regulated expression of mutant coding regions as well as to model loss-of-function (recessive) disease-associated alleles by inducible gene knockout (Fig. 2a, b).
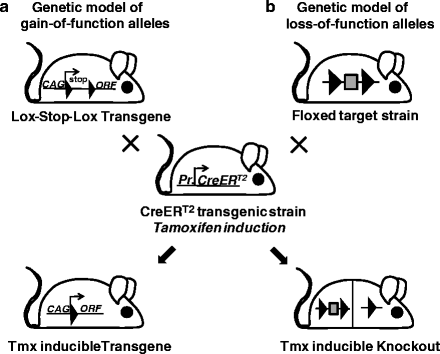
Fig. 2.
Tamoxifen inducible protein expression and gene knockout. (a) To obtain Tamoxifen inducible transgene expression, a mouse strain harbouring a loxP-flanked stop cassette between the CAG promoter and the open reading frame (ORF ) of interest (lox-stop-lox-transgene) is crossed to mice expressing CreERT2 fusion recombinase from a specific cell type promoter (Pr). In double transgenic offspring, transgene activation occurs in the selected cell type through Tamoxifen-induced Cre activation that deletes the stop cassette. (b) To generate Tamoxifen (Tmx) inducible knockout mice, a strain harbouring a loxP-flanked gene segment (floxed target strain) is crossed to mice expressing CreERT2 fusion recombinase from a specific cell type promoter (Pr). In double transgenic offspring, gene inactivation occurs in the selected cell type through Tamoxifen-induced Cre activation that deletes the floxed gene segment. LoxP sites are shown as filled triangles.
Using CreERT2 as a Tamoxifen-regulated switch, inducible transgenic mice can be conveniently generated by the insertion of a loxP-stop-loxP transgene into the Rosa26 locus of ES cells (Fig. 2a). The targeted insertion of the conditional expression unit into the Rosa26 locus ensures reproducible transgene expression that can be activated at any time in a cell type of interest. The expression vector for the protein of interest contains the respective open reading frame (ORF) located in between a loxP-flanked transcriptional stop element and a polyadenylation (pA) signal. The presence of the stop element initially prevents the production of mRNA containing the ORF. By the cross of lox-stop-lox transgenic mice with a CreERT2 strain (Fig. 2a), the ORF-coded protein becomes expressed from the vector-based CAG promoter upon the Tamoxifen-induced, Cre-mediated excision of the loxP-flanked stop cassette. Thereby the production of the protein of interest is controlled in vivo in a cell type-specific manner, depending on the chosen CreERT2 mouse line, and in a temporal manner by the administration of Tamoxifen at a chosen point in time. The inducible activation of Rosa26-based conditional transgenes has been used for the activation of reporter genes to characterise the recombination profile of CreERT2 transgenic lines (22–28) and for the forced expression of endogenous proteins (29, 30). By replacing reporter gene expression with coding regions for disease-associated proteins, this approach represents a very convenient and straightforward way for the generation of inducible transgenic mice. In this chapter, Sect. 3.1 covers the design of such Tamoxifen inducible transgene constructs. We provide a single step cloning approach for the generation of conditional vectors and a recombinase-based protocol for the simple vector insertion into the Rosa26 locus of ES cells.
Besides the activation of conditional transgenes, the Tamoxifen inducible CreERT2 gene switch is often used for gene inactivation in conditional knockout mice (Fig. 2b) (19–21). For this purpose, two loxP sites are placed into intron regions of the target gene such that they flank an essential coding exon but do not interfere with gene function. To enable inducible gene knockout, the floxed target strain is crossed to a chosen CreERT2 mouse line. Upon Tamoxifen administration, Cre-mediated excision of the loxP-flanked region results in the in vivo deletion of the coding segment from the genome of CreERT2 expressing cells. Upon deletion of the target exon, the mutant gene codes for a non-functional protein either by loss of an essential domain or as result of a reading frame shift. In Sect. 3.2, we cover the design of premade conditional knockout alleles generated by the EUCOMM/KOMP large-scale mutagenesis projects. Since the number of conditional alleles available from these projects is continuously expanding, self-made vector constructions are no longer mandatory to obtain a conditional mouse mutant. However, we further provide a simple PCR-based approach for the generation of conditional targeting vectors applicable in any standard laboratory. Finally, Sect. 3.3 summarises the use of CreERT2 transgenic mice. We include an overview of the available brain-specific CreERT2 mouse lines, notes on the control groups for inducible knockout experiments and a protocol for Tamoxifen administration.
Gene targeting in other species than mice
Over the past two decades, the mouse has developed into the prime mammalian genetic model to study human biology and disease because methods are available that allow the production of targeted, predesigned mouse mutants. This reverse genetics approach that enables the production of germ line and conditional knockout mice by gene targeting relies on the use of murine embryonic stem (ES) cell lines. ES cell lines exhibit unique properties such that they are able, once established from the inner cell mass of a mouse blastocyst, to renew indefinitely in cell culture while retaining their early pluripotent differentiation state. Gene targeting in ES cells has revolutionised the in vivo analysis of mammalian gene function using the mouse as genetic model system (31). However, since germ line competent ES cell lines that can be genetically modified could be established only from mice, this reverse genetics approach is presently restricted to this rodent species. The exception from this rule is achieved by homologous recombination in primary cells from pig and sheep followed by the transplantation of nuclei from recombined somatic cells into enucleated oocytes (cloning) (32, 33). Since this methodology is inefficient and time-consuming, it does not have the potential to develop into a simple routine procedure.
Experiments in model systems have demonstrated that the frequency of homologous recombination of a gene targeting vector is strongly increased if a double-strand break is induced within its chromosomal target sequence. Zinc-finger nucleases (ZFNs) were developed as a method to apply the stimulatory power of double-strand breaks to sequences of endogenous genes, without the need to introduce an artificial nuclease recognition site. Using zinc-finger nucleases in the absence of a gene targeting vector for homology directed repair, knockout alleles were generated in mammalian cell lines and knockout zebra fish and rats were obtained upon the expression of ZFN mRNA in one cell embryos (30, 34, 35).
Furthermore, ZFNs were used in the presence of exogeneous gene targeting vectors that contain homology regions to the target gene for homology-driven repair of the double-strand break through gene conversion. This methodology has been applied to gene engineering in mammalian cell lines and gene correction in primary human cells (36–38). More recently, zinc-finger nucleases together with gene targeting vectors were further used to generate targeted mutants in fertilised oocytes of mice and rats (39, 40). Therefore, ZFN-assisted gene targeting in zygotes provides a new, ES cell independent paradigm to manipulate the genome of mammals and other vertebrates with unprecedented freedom. In the near future, it will be therefore possible to create gain- and loss-of-function alleles as genetic PD models in non-murine species, e.g. the rat.
2 Materials
2.1 Tamoxifen Inducible Gene Expression
2.1.1 Plasmids
1.
pEx-CAG-stop-bpA
2.
pCAG-C31Int
3.
pRosa26-5′probe
2.1.2 Cells
1.
ES cell line IDG26.10-3
2.
Feeder cells: G418 resistant murine embryonic fibroblasts
2.1.3 Primers
1.
Neo: (5′-GTT GTG CCC AGT CAT AGC CGA ATA G-3′)
2.
Pgk (5′-CAC GCT TCA AAA GCG CAC GTC TG-3′)
3.
Hyg-1 (5′-GAA GAA TCT CGT GCT TTC AGC TTC GAT G-3′)
4.
Hyg-2 (5′-AAT GAC CGC TGT TAT GCG GCC ATT G-3′)
5.
SEQfor (5′-CATTATACGAAGTTATACC-3′)
6.
SEQrev (5′-ATC ATT TAC GCA ATT CCG C-3′)
Plasmids and ES cells are available from the authors
2.2 Tamoxifen Inducible Gene Knockout
1.
Plasmid pEasyfloxII-DTA (submitted to Addgene by R. Kühn)
2.
Genomic BAC clones (e.g. from ImaGenes, http://www.imagenes-bio.de or Source BioScience, http://www.lifesciences.sourcebioscience.com)
3.
Bioinformatic software for sequence analysis and vector design (e.g. VectorNTI, CLC Sequence Viewer, or Geneious)
5.
Proofreading DNA polymerase, e.g. Phusion (New England Biolabs) or Herculase-II (Stratagene)
6.
DNA oligonucleotides as PCR primers
2.3 Inducible Cre Activity
Tamoxifen stock solution: for i.p. injection, a 10 mg/ml stock of tamoxifen free base is prepared. Suspend 1 g tamoxifen free base (Sigma #T5648) in 10 ml 100% ethanol. Add 90 ml of sunflower seed oil (Sigma #S5007) and stir the suspension for several hours until the tamoxifen crystals are completely dissolved. Store the stock solution in aliquots at −20°C for up to 4 weeks.
3 Description of Methods
3.1 Tamoxifen Inducible Gene Expression
This section describes a simple and straightforward protocol for the production of transgenic mice that harbour a Tamoxifen inducible expression cassette within the Rosa26 locus on chromosome 6. To generate an expression vector for the protein of interest, a DNA fragment containing the respective ORF is ligated in between the loxP-flanked stop cassette and the polyadenylation (pA) signal of the generic vector backbone of pEx-CAG-stop-bpA (Fig. 3a). The presence of the stop cassette (a puromycin resistance coding region followed by triple pA signals) prevents the production of mRNA containing the ORF. The ORF-coded protein becomes expressed from the vector-based CAG promoter upon the Cre-mediated excision of the loxP-flanked stop cassette. By this means, the production of the protein of interest can be controlled in vivo in a cell type-specific and temporal manner, depending on the characteristics of the chosen CreERT2 transgenic mouse line. The pEx-CAG-stop-ORF-bpA conditional expression vector (Fig. 3b) is inserted by recombinase-mediated cassette exchange (RMCE) into the modified Rosa26 allele of IDG26.10-3 ES cells (Fig. 3c).The targeted insertion of the vector into the Rosa26 locus ensures reproducible transgene expression that can be activated in any cell type of interest. For the stable integration into the ES cell genome, we use a RMCE protocol based on the attB and attP recognition sites of phiC31 integrase that we initially developed for the targeted insertion of shRNA expression cassettes into Rosa26 (41, 42). Figure 3b illustrates the C31Int/attB/attP-mediated cassette exchange at a pre-engineered Rosa26 docking site. Since the pEx-CAG-stop-ORF-bpA plasmid includes a promoterless neomycin resistance gene (neo), which is activated only after correct cassette exchange at the Rosa26 acceptor locus (Fig. 3d), RMCE positive ES cell clones can be conveniently selected in G418. Finally, RMCE ES cell clones are used for the generation of chimaeric mice and germline transmission of the conditional transgene.
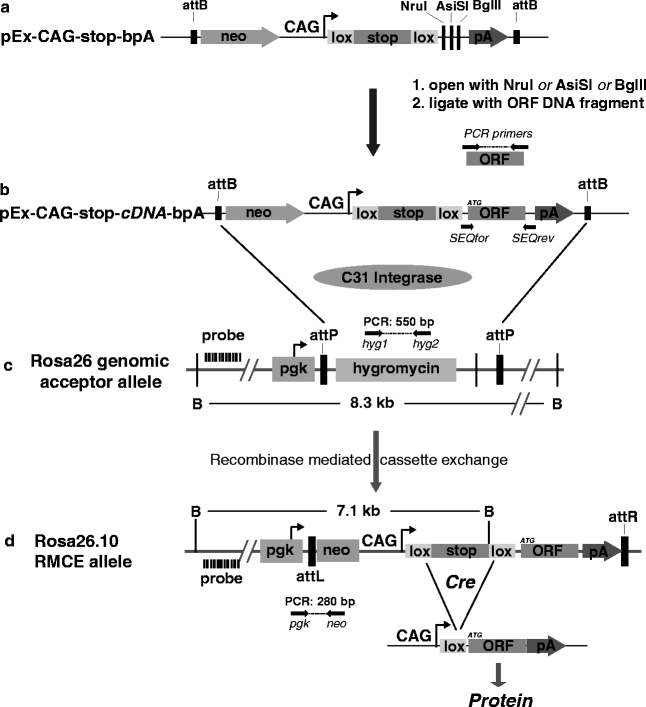
Fig. 3.
Construction of Tamoxifen inducible transgenes and integration into the Rosa26 locus of ES cells. (a) Schema of the conditional expression vector pEx-CAG-stop-bpA containing a promoterless neomycin resistance (neo) gene, the CAG promoter followed by a loxP-flanked transcriptional stop cassette, NruI, AsiSI and BglII restriction sites for the insertion of a DNA fragment containing an open reading frame (ORF) for protein expression and a polyadenylation (bpA) signal. These elements are flanked by attB recognition sites for phiC31 integrase. (b) Completed conditional expression vector pEx-CAG-stop-ORF-bpA obtained by ligation of an ORF into the NruI, AsiSI or BglII site of plasmid pEx-CAG-stop-bpA. Primers SEQfor/rev can be used for the sequence confirmation of the insert. For genomic integration, this vector is transfected together with a C31 integrase expression plasmid into Rosa26.10 ES cells harbouring a modified Rosa26 allele containing two C31 integrase attP recognition sites. (c) The Rosa26 acceptor locus in IDG3.2 Rosa26.10 ES cells contains a pgk promoter and a hygromycin resistance coding region flanked by two attP recognition sites. PCR genotyping can be performed with the primer pair hyg1/hyg2 resulting in a 550 bp PCR product. Using the Rosa-5′ genomic hybridization probe and BamHI (B) digestion, the acceptor allele is recognised as a 8.3 kb band. (d) Upon C31 Integrase-mediated recombination of both pairs of attB and attP sites, the hygromycin resistance gene becomes exchanged by recombinase-mediated cassette exchange (RMCE) against the ORF expression unit. ES cell clones habouring RMCE alleles can be selectedby Fig. 3. (continued) the neo resistance gene that is expressed from the pgk promoter within Rosa26. Recombined ES cell clones can be identified by PCR genotyping using the primer pair pgk/neo that amplifies a 280 bp fragment. Using the Rosa-5′ genomic hybridization probe and BamHI (B) digestion, the RMCE allele is recognised as a 7.1 kb band, in contrast to the second Rosa26 wildtype allele that generates a 5.8 kb BamHI fragment (data not shown). Upon the Cre-mediated excision of the loxP-flanked stop cassette in vivo, the ORF is transcribed and translated into the desired protein.
3.1.1 Cloning of Inducible Expression Vectors
1.
Digest DNA of plasmid pEx-CAG-stop-bpA with the single cutters NruI, AsiSI, BglII or a combination of these enzymes and dephosphorylate the open ends. Generate a PCR-based DNA fragment containing the ORF of interest. Include into the PCR primers appropriate restriction sites for ligation into the pEX-CAG-stop-bpA plasmid and add, immediately upstream of the ATG start codon, a Kozak consensus sequence (GCCACC) to ensure efficient translation. Ligate 50 ng of the digested and purified PCR fragment with 100 ng opened pEx-CAG-stop-bpA in a 15 μl standard ligation reaction.
2.
Transform the ligation reaction into Escherichia coli cells (we use DH5α), and digest miniprep DNA with a combination of ORF internal and external (NruI, AsiSI, BglII) restriction enzymes to determine the presence and orientation of the insert.
3.
Prepare Maxiprep (100 ml LB culture, Quiagen Plasmid Maxi kit, Quiagen) with one confirmed clone of pEx-CAG-stop-ORF-pA and use primers SEQfor/rev for the sequence verification of the ligated insert.
3.1.2 Stable Vector Integration into the Rosa26 Locus of ES Cells by RMCE
1.
Co-electroporate 25 μg (circular) DNA of pEx-CAG-stop-ORF-pA together with 25 μg (circular) DNA of the C31 Integrase expression vector pCAG-C3Int into IDG26.10-3 ES cells grown on feeder layers. For electroporation of 50 μg plasmid DNA, use 4 × 106 ES cells in 0.8 ml PBS with a 0.4 cm BioRad cuvette (we use the BioRad Genepulser Xcell at 300 V and 2 ms time constant). Plate transfected cells into two 10-cm culture dishes containing embryonic fibroblasts as feeder layer.
2.
On day 2 after transfection, add G418 (140 μg/ml) to the culture medium. Keep in selection for 8 days by changing medium daily.
3.
On day 9: pick 12 (or more) colonies per RMCE experiment. Expand cells on feeders for freezing and on gelatine for DNA isolation.
4.
Extract genomic DNA by standard procedures (43) and identify positive clones by PCR or Southern blotting as described below.
3.1.3 Identification of RMCE Positive ES Cell Clones
1.
For PCR identification of RMCE positive ES cell clones use the neo and pgk primers listed in Sect. 2.1 (annealing temperature: 65°C; correct product size 280 bp). Note that feeder cells containing a pgk-neo resistance gene will yield an additional amplification product of 160 bp. Incorrectly recombined clones will still contain the hygro gene which can be checked by using the Hyg-1 and Hyg-2 primers listed in Sect. 2.1 (annealing temperature 65°C, product size 550 bp). Note that using feeders harbouring an additional hygro resistance gene precludes this screening procedure.
2.
Southern blot verification of correct clones is mandatory for exclusion of partially recombined alleles or chromosomal rearrangements. To distinguish the wildtype and RMCE Rosa26 alleles, cleave the genomic DNA with BamHI and use the 450 bp EcoRI fragment from plasmid pRosa-5′probe as hybridization probe. The wildtype Rosa26 locus is identified by a band of 5.8 kb. The parental IDG26.10-3 ES cells show an additional band of 8.3 kb derived from the modified Rosa26 allele before RMCE (Fig. 3c). Positive RMCE clones show a new band of 7.1 kb due to the integration of the neo and CAG-stop-ORF gene cassettes (Fig. 3d). Usually, ∼40% of the neomycin resistant clones undergo complete RMCE.
3.
Use confirmed clones for generating transgenic mouse lines by blastocyst injection or tetraploid embryo aggregation as described in (44).
3.2 Tamoxifen Inducible Gene Knockout
Conditional mutagenesis enables inactivation of a target gene via Cre/loxP recombination in somatic cells. In the conditional allele, a functionally essential (critical) exon is flanked by two loxP sites that do not interfere with gene function (Fig. 4c, 5e). Conditional mutant mice are obtained by crossing the conditional strain with a transgenic line that constitutively expresses Cre recombinase or the Tamoxifen inducible CreERT2 fusion protein under control of a cell type-specific promoter. Thereby, the inactivation of the target gene is either restricted to a selected cell type without temporal control or can be induced at a chosen time.
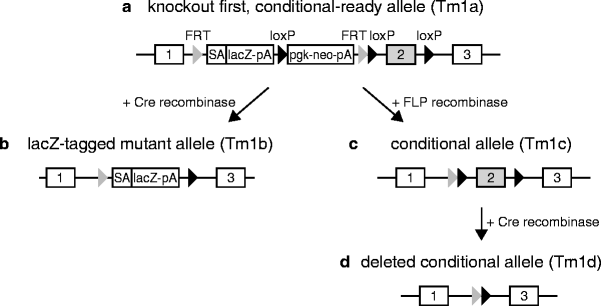
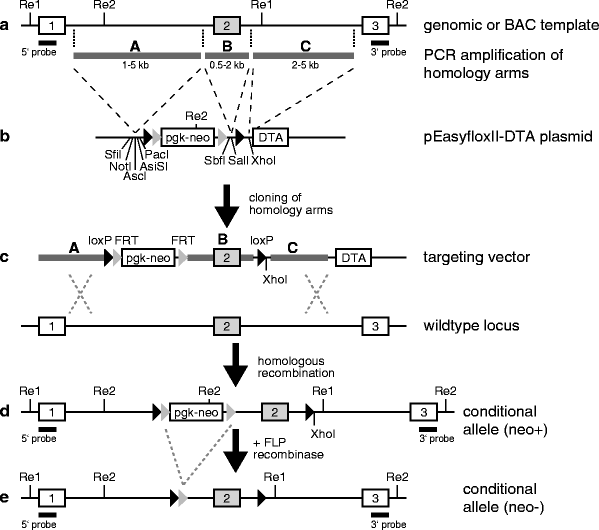
Fig. 4.
Gene targeting using “knockout first, conditional-ready” alleles. The “knockout first, conditional-ready” allele (Tm1a, a) consists of a β-galactosidase reporter cassette (“lacZ-pA”) and a neomycin expression cassette (“pgk-neo-pA”) flanked by two FRT sites. The critical exon 2 is flanked by two loxP sites, a third loxP site is located between the lacZ and neo genes. Upon Cre recombination of the Tm1a allele, exon 2 and the neomycin cassette are excised, leading to the lacZ-tagged knockout allele (Tm1b, b). If the knockout first allele is initially recombined using FLP recombinase, the β-galactosidase cassette becomes deleted, creating a conditional knockout allele (Tm1c, c). Subsequent recombination with Cre recombinase in specific cell types leads to the conditional inactivated allele (Tm1d, d). Exons of the target gene are shown as numbered rectangles.
Fig. 5.
Targeting vectors for conditional knockout alleles. (a) Genomic DNA or a BAC clone is used for the PCR amplification of the targeting vector’s homology arms A, B and C, which are subsequently ligated into the backbone of the pEasyfloxII-DTA plasmid (b). (c) The resulting targeting vector is used for the mutagenesis of the target gene by homologous recombination, leading to a neomycin-positive mutant allele (d). Finally, the neomycin cassette is excised using FLP recombinase to obtain the conditional allele (e). Exons of the target gene are shown as numbered rectangles.
Due to the substantial existing and growing sources of information on targeting vectors, ES cell clones, and mutant mouse strains, a mutagenesis project should be started by searching the relevant databases for preexisting materials to avoid potential duplication. This can be conveniently achieved by searching for the name of the gene of interest at the MGI (http://www.informatics.jax.org), IGTC (http://www.genetrap.org) and IKMC (http://www.knockoutmouse.org) Web pages. If these databases do not yield materials relevant to the project requirements, it may be necessary to consider generating a novel conditional gene targeting vector, as described in Sect. 3.2.2.
3.2.1 Conditional Alleles Generated By Large-Scale Mutagenesis Programmes
The large-scale EUCOMM/KOMP mutagenesis programmes, which are coordinated by the International Knockout Mouse Consortium (IKMC), generate a genome-wide resource of gene-specific targeting vectors, targeted ES cell clones and mutant mouse strains. The EUCOMM/KOMP alleles are mostly of the “KO first, conditional-ready” type (Fig. 4) (45). This design initially disrupts the target gene by the intronic insertion of a gene disruption cassette that includes a splice acceptor element, a β-galactosidase reporter gene and a neo resistance gene. In these “knockout first” alleles, gene function is inactivated by splicing of upstream exons to the splice acceptor site of the targeting cassette (Targeted mutation 1a, Tm1a, Fig. 4a). Most EUCOMM/KOMP targeting vectors contain a targeting cassette with a neo resistance that is driven by its own β-actin promoter to allow the targeting of all genes, irrespective of their expression status in mouse ES cells. Promoterless vectors that utilise the promoter of the targeted gene to drive the expression of the neo resistance are also used for the targeting of genes that are expressed in ES cells. In addition to the gene disruption cassette, the targeted EUCOMM/KOMP alleles contain two loxP sites that flank a critical exon of the target gene (Fig. 4a). The loxP-flanked critical exon, as the target for Cre-mediated excision, is determined by these criteria: (1) its deletion causes a translational reading frame shift in the remaining mRNA that leads to the production of a shortened, mutant protein, (2) the exon is present in all transcript splice variants of the targeted gene, (3) the size of the exon is less than 1 kb, to ensure that the distance of the loxP sites is minimal for efficient Cre recombination, (4) the size of the flanking introns is at least 0.5 kb, so that loxP sites can be placed in non-conserved regions that are not required for endogenous splicing. In many cases, the second exon of a target gene fulfils these critical criteria, but in some instances several small exons are combined into a group of critical exons.
The “KO first,” Tm1a allele can be converted into a classical KO allele by breeding the Tm1a mice with a Cre transgenic germline deleter strain. By this means, the neomycin selection cassette and the critical exon are deleted in the germline, resulting in a β-galactosidase-tagged, classical KO allele (Tm1b, “beta-Gal reporter,” Fig. 4b). Furthermore, the Tm1a-type allele can be converted into a Tm1c allele for conditional mutagenesis by the use of the two FLP recombinase recognition (FRT) sites that are flanking the gene disruption cassette. The Flp-mediated deletion is typically performed by breeding the Tm1a mice with an Flp transgenic germline deleter strain (for a line in the C57BL/6N background, see ref. 46). Upon the removal of the gene disruption cassette, the remaining loxP sites that are flanking the critical exon delineate the configuration of a conditional allele (Tm1c, “conditional,” Fig. 4c). This functional conditional allele can subsequently be inactivated in somatic cells by breeding Tm1c mice to a mouse line that expresses Cre recombinase in a time- and tissue-specific manner, resulting in the non-functional Tm1d allele (“deletion,” Fig. 4d). At present, the EUCOMM/KOMP resource offers >10,000 targeting vectors, >8,000 genes targeted in the C57BL/6N-derived ES cell line JM8 (47) and >700 established mouse strains. These resources are accessible through the programmes in the Web pages (http://www.eucomm.org; http://www.komp.org) and the IKMC (http://www.knockoutmouse.org).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


