Chapter 16 Fluids, Electrolytes, and Acid–Base Therapy
Physiology
Water
Water accounts for 60% of lean body weight in adult animals, with variation between species. This percentage is affected by fat and age. Obese and older animals tend to have a smaller percentage of body weight composed of water, whereas up to 90% of body weight in neonates is water. Approximately two thirds of this water is present within cells (intracellular fluid [ICF] compartment) and one third is extracellular fluid (ECF). Of the ECF, three fourths is interstitial fluid and one fourth of the ECF volume is plasma. Thus plasma accounts for only 5% of body weight.1,2
Water freely diffuses through cell membranes. Consequently, all body compartments have approximately the same osmolality (290 to 310 mOsm/kg in dogs and 300 to 330 mOsm/kg in cats).3 Osmotic force is the prime determinant of distribution of water across cell membranes (i.e., the partitioning of water between the ECF and ICF). Plasma sodium is the primary determinant of ECF osmolality. Glucose and urea make minor contributions as well, which is reflected in the following calculation:
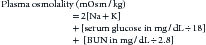
As can be seen from the preceding formula, sodium is principally responsible for ECF osmolality. Osmolality in the ICF is principally determined by potassium, magnesium, phosphates, and proteins. To promote water movement from one compartment to another, the osmolality of one or more compartments must change.4 Tonicity, or effective osmolality, refers to the osmotic pressure of a solution—Osmotic pressure is the pressure required to prevent water movement across a semipermeable membrane. When the relationship between tonicity and osmolality is under consideration, it is important to distinguish between permeant and impermeant solutes. Permeant solutes (e.g., urea) move freely across cell membranes and do not induce net water movement when introduced into a solution (i.e., they are ineffective osmoles). Impermeant solutes (e.g., sodium and glucose) induce water movement when introduced into a solution and are called effective osmoles, because they do not readily cross cell membranes. Glucose, to the extent that it causes hyperglycemia, is an impermeant solute and does contribute to ECF osmolality. However, glucose administration usually does not cause hyperglycemia because it is rapidly taken up by cells in the presence of insulin. The implication is that tonicity is less then osmolality.
The capillary walls are freely permeable to sodium, chloride, and glucose. As a result, these substances are osmotically inactive across capillary membranes. However, plasma proteins are limited in their ability to cross capillary membranes, and plasma volume is ultimately maintained by the colloid osmotic pressure (COP), COP is the force created by macromolecules present within the vasculature that prevents water from escaping to the extravascular space. Thus, COP is essentially the pressure exerted by plasma proteins. Albumin contributes to roughly 75% of COP in healthy patients, with globulins making up the remainder. At the venous end of the capillary bed, plasma proteins exert an osmotic force in excess of the hydrostatic gradient, resulting in a net fluid flux from the interstitium into the vessels. Thus albumin is important for distribution of water between the intravascular and interstitial compartments, and plasma proteins in general play a key role in the maintenance of intravascular fluid volume.1 This is in contrast to the osmotic equilibrium governing water distribution across the ECF and ICF compartments.
The amount of available water in the body is determined by a balance between intake and loss.1 Water intake occurs by drinking and eating, which are controlled by thirst and hunger. Alterations in water intake can occur with neurologic disease and congenital abnormalities. Animals normally lose water through urine, feces, and expired air. To a lesser extent, evaporation from the skin surface promotes water flux across the epidermis. Obligatory urinary and fecal losses are determined by the solute load that must be excreted. Respiratory losses of water are affected by ambient temperature and humidity. Water losses may increase as a result of vomiting, diarrhea, and polyuria. Rarely, clinically relevant water losses arise through traumatized tissues or with cavitary effusions. Fever also can be responsible for increasing loss of water.5 Fluid losses, such as those incurred by vomiting, diarrhea, and polyuria, are often classified as isotonic (i.e., small solutes are lost in proportion to their concentration in plasma), but more commonly these losses are slightly hypotonic, with sodium concentrations of 80 to 120 mg/dL. This is manifested by hypernatremia, which is commonly observed with dehydration. To maintain hydration homeostasis, urine output may exceed the obligatory urine output, the latter being the volume required for elimination of solutes. While the term free water is well recognized and accepted, this fluid resembles a hypotonic crystalloid in content, as opposed to water. Water excretion is due to modification of antidiuretic hormone (ADH) secretion. ADH is normally released from the posterior pituitary gland when osmoreceptors in the hypothalamus detect hypertonicity or when baroreceptors in the cardiovascular system sense hypovolemia. Release of ADH promotes water retention. For water to be excreted by the kidneys, there must be adequate delivery of tubular fluid to the ascending limb of the loop of Henle, and the renal collecting ducts must remain impermeable to water in the absence of ADH.6
Sodium
Sodium is the osmolar skeleton of the ECF. Under normal conditions the kidneys regulate its elimination, excreting as much sodium as is ingested. Renal sodium excretion is normally regulated by aldosterone, atrial natriuretic factors (ANF), and intrinsic renal mechanisms (e.g., renal blood flow [RBF], glomerular filtration rate [GFR], and glomerulotubular balance). Glomerulotubular balance refers to the ability of the kidney to maintain a relatively constant fractional reabsorption of sodium, despite changes in GFR. If there is expansion of the effective circulating volume (i.e., overhydration, hypervolemia), the cardiac atria distend with subsequent release of ANF. ANF and other natriuretic factors inhibit both the formation and effects of angiotensin and decrease sodium reabsorption from the renal medullary collecting ducts. Conversely, when atrial receptors detect a decrease in volume, sympathetic tone is increased and ANF release is inhibited. Further, decrements in renal perfusion and the associated decrease in delivery of sodium (and/or chloride) to the macula densa activate the renin–angiotensin–aldosterone system, promoting sodium to be reabsorbed from the renal tubules and collecting ducts.7
Potassium
The regulation of total body and plasma potassium concentration is important to the extent that potassium alterations may profoundly affect the resting membrane potential (RMP) of cardiac and neuromuscular cells.8 Hypokalemia lowers the RMP, making it more difficult to achieve an action potential with subsequent contraction of a muscle. Hyperkalemia raises the RMP, which may result in an action potential of decreased amplitude or, in extreme cases, continuous depolarization of the cell membrane. Although the majority of the body’s potassium is found within cells, it is the plasma concentration that corresponds with and contributes to relevant myocardial and neuromuscular abnormalities.
Potassium homeostasis can be conceptualized as having both an internal and an external balance. External balance refers to the total amount of potassium in the body and is determined by the balance between intake versus loss. Potassium enters the body principally through ingestion, and almost all ingested potassium is absorbed. A small amount of potassium is normally excreted in the feces, although the colon may serve as an important route for potassium excretion in disease states. Normal potassium loss occurs predominantly through the kidneys, the amount determined by ECF potassium concentration, delivery of sodium and water to the distal tubule and collecting ducts, as well as the secretion of aldosterone. Although the kidneys normally eliminate potassium efficiently, renal mechanisms cannot maintain normal plasma serum potassium concentrations when grossly excessive amounts of potassium are ingested.9
Internal potassium balance refers to the distribution of potassium between the ICF and ECF compartments. The Na-K ATPase enzyme pump found in cellular membranes actively transports potassium into the cell and maintains a high intracellular potassium concentration (i.e., approximately 150 mEq/L) relative to the ECF (approximately 4 mEq/L). In hyperkalemic states insulin and β2-adrenergic activity augment the transport of potassium into the cells, safely storing additional potassium until it can be eliminated.9 The internal balance mechanisms may be thought of as a temporary measure designed to allow the kidneys and, to a lesser extent, the colon time to restore potassium balance.8,10
Acid–Base Balance
The body must deal with very large amounts of acid (H+) that are generated daily by normal metabolic processes. This is important because H+ is very reactive, and small amounts (i.e., nanoequivalents) can have detrimental effects on protein structure and function (i.e., enzymes, cell membranes, and receptors). Normal blood pH is 7.40. Many metabolic functions are exquisitely sensitive to pH, and normal function can only occur within a very narrow pH range. If, for example, a patient’s pH falls to 7.20 (which represents an increase of approximately 20 nEq H+/L), myocardial dysfunction, vasodilation, or dysrhythmias may be observed.11,12
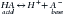
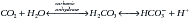
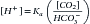
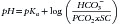
There are two categories of acid found in the body: nonvolatile (produced by metabolism of proteins) and volatile (derived from CO2) produced by cellular respiration throughout the body.11 Nonvolatile (also called fixed) acid (H+) is primarily excreted by the kidneys, whereas volatile acid (CO2) is eliminated by way of the lungs. On a daily basis, small pH changes within the body are counteracted by multiple complex and often opposing physiochemical processes that, for the sake of simplicity, are presented as (1) the actions of intracellular and extracellular buffering systems (chemical buffering), (2) modulation of ventilation (physiologic buffering), and (3) renal reclamation and elimination. There are many buffer systems throughout the body. The principle intracellular buffers are hemoglobin, phosphate, and proteins, which are capable of buffering both volatile and nonvolatile acids. The principle extracellular buffer systems are calcium carbonate and phosphate of bone, in addition to the bicarbonate-carbonic acid system, which reacts only with nonvolatile acid. Hemoglobin and albumin account for most of the nonbicarbonate buffering capacity. The HCO3− buffering system is capable of responding to an acute change in [H+], and the HCO3−/H2CO3/CO2 equilibrium equation (see Equation 16-1) allows changes in pH to be further modulated by changes in ventilation. This “open system” greatly enhances the buffering capacity of the HCO3− system and is capable of buffering changes in pH within minutes of an acid or alkali load. Finally, the kidneys play a major role in maintaining pH by increasing or decreasing acid elimination in the urine. Although hours to days may be required for this system’s buffering ability to reach completion, it is clinically the most reliable of all the adaptive mechanisms to normalize pH.
Acid–base physiology may be viewed as having an external and an internal balance, as described for potassium. External balance refers to the elimination of acid from the body, whereas internal balance allows for the body to safely sequester excessive acid until it can be excreted. The pH of the ECF is largely determined by how effectively these mechanisms function.
A perceived limitation to traditional acid–base physiology is that it is too simplistic, ascribing changes in pH to either respiratory (changes in pCO2) or metabolic (changes in bicarbonate) causes, thereby neglecting the contribution of other variables in solution. To address this, nontraditional or quantitative approaches to acid–base chemistry and analysis are described.13 The terms Stewart approach and Fencl–Leith approach are variants of the often confusing and misunderstood nontraditional approaches to acid–base analysis. The nontraditional approach subdivides metabolic causes into two independent variables, those associated with a strong ion difference (SID) ions that are totally dissociated (e.g., sodium, chloride, potassium, lactate) and ATOT (ions of weak acids that are partially dissociated, e.g., phosphate and albumin). The nontraditional approach defines the pCO2 (respiratory contribution) as an independent variable. According to the quantitative approach, changes in HCO3 are explained by alterations in SID, ATOT, and pCO2. It has been demonstrated that if enough ATOT and SID determinants are measured, an approximate pH can be calculated. The perceived strength of the quantitative approach is that it provides an estimate of the “contribution” of individual electrolyte derangements to an acid–base imbalance when pH is not measured. However, the quantitative approach is only a means of estimating the metabolic contribution to pH when it is not otherwise measured, but not the pH itself; the pH is also highly dependent on the respiratory component, the pCO2. The traditional system utilizes an overview parameter such as bicarbonate, total CO2, or base deficit to quantify the metabolic contribution. The Stewart approach breaks down the metabolic component into two large groups: SID and ATOT; however, it does not further define the contributions. The Fencl–Leith modification is the only truly semiquantitative approach, insofar as it it breaks down the metabolic contribution into six individual components (water [marked by serum sodium], bicarbonate [marked by chloride], albumin, lactate, ketones [if measured], phosphate, and the effect of unmeasured ions). Advocates of the nontraditional approach find that it is more accurate, avoiding some of the oversimplifications associated with the traditional, albeit more familiar, Henderson–Hasselbalch equation. Both are simplifications of a complex dynamic system, and both suffer from inherent inaccuracies.13 For practical purposes, benefits of the nontraditional approach are best appreciated when pH and a blood gas analyzer are not available to the clinician. Other advantages to this approach, when compared with traditional acid–base chemistry, are less clear and remain a topic of recurrent deliberations. Importantly the astute clinician should appreciate that all methods of acid–base analysis merely quantitate the magnitude of a disorder. Similarly, acid–base findings may provide subtle clues to pathogenetic mechanisms, but they do not characterize or define a disease or its management. Such inferences mandate evaluation of the complete history, thorough physical examination, and results of other laboratory tests. To remain consistent with concepts taught in most clinical settings, the traditional approach (Henderson–Hasselbalch) to analyzing acid–base disturbances is discussed.
When nonvolatile acid is present in excess, it reacts with bicarbonate and other buffer systems, especially intracellular protein and phosphate.14 The plasma bicarbonate concentration decreases as the bicarbonate combines with H+ and forms H2CO3, the pH decreases, which by definition is an acidosis. More specifically, it is a metabolic acidosis because the disorder promotes the accumulation of excessive nonvolatile acid. The body will try to reestablish pH by lowering the pco2 (hyperventilation). This is the appropriate and anticipated compensatory response. Although less common, excessive loss of HCO3− (as occurs with some types of diarrhea) may cause a metabolic acidosis with a similar compensatory response.15 When the primary derangement promotes accumulation of excess volatile acid (an increase in pCO2), pH decreases and the acid–base disorder is described as a respiratory acidosis. Respiratory acidosis, by definition is an increase in pCO2,. In patients with primary respiratory acidosis, the kidneys respond by increasing excretion of acid.15
By definition, metabolic alkalosis is an elevation in pH caused by excessive plasma bicarbonate. Alkalosis decreases pulmonary ventilation, which elevates the pco2 Conversely, reduction of pco2 produces an alkalosis, the terms hyperventilation and respiratory alkalosis often used interchangeably. In response to a primary respiratory alkalosis, renal acid excretion is reduced.16
The kidneys respond to respiratory acid-base disorders over several hours to days. The pulmonary response to metabolic acid-base disorders occurs in a matter of minutes. In dogs it is possible to predict the approximate degree of a compensation; anticipated compensation is less clear or predictable in cats. Compensatory mechanisms are not efficient enough to return the pH within the normal range. These defense mechanisms do not correct the acid-base disturbance but merely minimize the change in pH imposed by the disturbance. Moreover, overcompensation for a primary acid–base disorder does not occur,11
Disease-Induced Changes
Sodium
Changes in plasma sodium concentration usually reflect changes in total body water content. Hyponatremia and hypernatremia primarily cause clinical problems by promoting neuronal edema or dehydration (ICF), respectively. The severity of clinical signs is thought to correspond with the rate of change rather than with the magnitude of hyponatremia or hypernatremia. If changes in serum sodium occur slowly, the brain can usually adjust the number of osmotically active molecules to prevent cerebral fluid gain or loss. This underscores the fact that severe hyponatremia or hypernatremia should be corrected gradually rather than quickly.17 When the serum sodium abnormality has been gradual in onset, the patient’s sodium should be corrected slowly, not lowered faster than 1 mmol/L/hr or increased at a rate exceeding 0.5 mmol/L/hr.
Hypernatremia
Naturally occurring disease processes resulting in hypernatremia are usually disorders of free water loss.7,18 Rarely, hypernatremia results from the addition of sodium (e.g., ocean water ingestion, hypertonic saline or sodium bicarbonate administration). Hypernatremia is a reflection of inadequate water relative to sodium content in the ECF. While ECF volume is determined by the total sodium content (not concentration) in the body, plasma sodium concentration is not a measurement of a patient’s volume status. The thirst mechanism is so effective that hypernatremia seldom occurs in animals that have access to adequate amounts of water and are not vomiting or regurgitating. Hypernatremia often develops in hospitalized patients, and a methodical preemptive diagnostic approach, including serial evaluation of electrolytes and quantification of fluid intake and loss, permits rapid recognition of water imbalance and prevent wide variations in serum sodium. Hypernatremia should prompt consideration of the disorders listed in Box 16-1.
Hyponatremia
Hyponatremia may be caused by several mechanisms, and a systematic approach to cause and correction is advised. Sodium and its attendant ions account for approximately 95% of the osmotically active substances in extracellular water. While hyponatremia is commonly associated with hypo-osmolality, plasma osmolality must be assessed to appropriately determine of the cause of hyponatremia (Box 16-2).7
Box 16-2 Evaluation of the Patient with Hyponatremia
Hyponatremia with concurrent normal plasma osmolality suggests laboratory error or pseudohyponatremia. Pseudohyponatremia may occur with hyperlipidemia because the lipid occupies space in the volume of serum obtained for analysis; the concentration of sodium itself is not affected. Hyponatremia with concurrent plasma hyperosmolality suggests that other osmotically active particles (e.g., glucose, mannitol) are drawing water out of the ICF and into the plasma, thus diluting the sodium that is present.7,19 This is often called translocational hyponatremia because it is caused by translocation of sodium across cell membranes.
Volume and hydration status may further suggest causes of hyponatremia. Overtly hypovolemic patients often have an obvious source of fluid loss. Hyponatremia in an overhydrated patient is often observed with (but is not the cause of) cavitary effusions associated with congestive heart failure, advanced oliguric renal failure, or severe hepatic disease (usually cirrhosis). These disorders may be associated with such exuberant water retention that patients become hyponatremic in the face of total body sodium excess. Patients with congestive heart disease or cirrhosis are often described as having decreased effective circulating volume (ECV). ECV is the volume of arterial blood effectively perfusing tissue. ECV is a dynamic quantity and not a measurable, distinct compartment. The fluid retention seen with ineffective cardiac output is appropriate (because of release of ADH and angiotensin II), whereas the fluid retention observed with renal failure and cirrhosis is a component of the disease process itself. These patients have decreased ECV and consequent reduction in GFR and RBF; as a result, the body attempts, albeit ineffectively, to retain the fluid within the ECF.7
Hypoosmolar hyponatremia may be seen with chronic gastrointestinal disease. These patients may be hypernatremic early in the course of their disease, but sustained ADH release, excessive water consumption, or both may ultimately lead to hyponatremia. Diseases associated with third-space losses (e.g., pancreatitis, peritonitis) promote ADH release, and hyponatremia may be seen in affected patients. It is a common misunderstanding that hyponatremia in this setting is a result of the fluid accumulation itself. The hyponatremia, however, is due to changes in ADH, and fluid accumulation is not the proximate cause. Cutaneous fluid losses (e.g., burns) may be isonatremic with little change in plasma sodium or may be associated with hyponatremia.7,19
Under normal circumstances, the body’s response to hypovolemia and hyponatremia is to impede renal sodium losses as a means of restoring ECF. Consequently, little sodium would be found in the urine (FE Na+ <1%). Animals in which renal losses are the cause of the hyponatremia will, however, have substantial urinary sodium concentrations (FE Na+ >3%).19
Hyponatremia and hypoosmolality occur uncommonly in well-hydrated euvolemic dogs and cats. Although rare, primary polydipsia (also called psychogenic polydipsia) syndromes associated with inappropriate ADH release or overzealous administration of hypotonic fluids are described, and are possible causes of hyponatremia in this setting.7
Chloride
Most often, changes in plasma chloride concentration are clinically relevant because they affect acid-base status. In particular, hypochloremia is associated with metabolic processes that are coupled with alkalosis.20 Similarly, hyperchloremia tends to be associated with acidosis.
Hypochloremia
The major causes of hypochloremia are increased loss caused by vomiting of gastric contents or excessive administration of loop diuretics (e.g., furosemide).21 Chloride losses caused by gastric vomiting are usually associated with hypokalemia and metabolic alkalosis. Occasionally, a paradoxical aciduria is also seen. Physical examination and laboratory findings often corroborate a history of vomiting. Rarely, administration of large doses of sodium without corresponding administration of chloride (e.g., high doses of sodium penicillin or sodium bicarbonate) may cause hypochloremia. When a hypochloremic patient is also hyponatremic, hyperkalemic, or both, the clinician should consider hypoadrenocorticism. If, however, the clinician corrects the plasma chloride concentration to account for changes in plasma free water, for example:
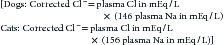
patients with hypoadrenocorticism may have a normal or increased corrected chloride concentration.20 Changes in chloride are often interpreted with changes in free water, which alters sodium and chloride concentrations proportionally. This can be done by correcting the chloride concentration for changes in sodium using the preceding formulas. Primary disorders of chloride (i.e., acid–base disturbances) have abnormal corrected chloride values, whereas disorders of free water do not; determination of corrected chloride values seldom affects diagnostic or therapeutic decisions in a given patient. Its determination is only to permit the interested clinician to quantitatively assess the contribution of bicarbonate (marked by changes in chloride) to the metabolic component of an acid–base disturbance.
Hyperchloremia
Hyperchloremia is principally found in animals that are simultaneously hypernatremic as a result of the loss of free water and in patients that have received fluids containing proportionally more chloride than sodium relative to plasma values. Examples include administration of 0.9% saline, hypertonic saline, parenteral nutrition, and especially 0.9% saline supplemented with potassium chloride. Hyperchloremia may also be observed in patients that have a metabolic acidosis associated with a normal anion gap (i.e., hyperchloremic metabolic acidosis). This biochemical finding is commonly seen in animals with small bowel diarrhea caused by loss of bicarbonate-rich, chloride-poor fluid.20 It can also be seen with regurgitation or vomiting of duodenal contents and in patients with renal tubular acidosis.
Potassium
Changes in the plasma potassium concentration are clinically important because of the effect of potassium on cellular metabolism, RMPs, and the subsequent strength of contraction when an action potential is generated.9 Hypokalemia and hyperkalemia both cause muscular weakness and predispose to cardiac dysrhythmias. Although there is variation between patients, plasma potassium concentrations ≥ 8.0 mEq/L or ≤ 2.0 mEq/L are generally considered life threatening, although less dramatic changes can be dangerous if there are concomitant electrolyte derangements (e.g., hyponatremia, hypocalcemia, or hypercalcemia) or if the changes occur rapidly.10 Potassium is highly labile, and changes in pH of the ECF may cause significant alterations in serum potassium, particularly in critically ill patients.
Hyperkalemia
Hyperkalemia may be artifactual (i.e., pseudohyperkalemia) or real. If hyperkalemia is real, it is either iatrogenic (increased intake or administration) or spontaneous. The major cause of spontaneous hyperkalemia is decreased urinary excretion caused by renal, urinary tract or adrenal disease (Box 16-3).10
Box 16-3 Causes of Hyperkalemia
Artifactual
Pseudohyperkalemia, an in vitro increase in potassium concentration, may be observed with marked thrombocytosis (generally ≥ 800,000/ul). Because the elevation in potassium in this setting is due to leakage from disrupted or dying cells, it is important to measure plasma or serum potassium concentrations shortly after a sample is obtained to avoid drawing erroneous conclusions about a patient’s electrolyte status. Similarly, animals with white blood cell counts equal to or greater than 100,000/ul (e.g., leukemia) may rarely have pseudohyperkalemia because of transcellular leakage of potassium.22,23 Neonates, Akitas, and English springer spaniels are known to have a high potassium content within their red blood cells and hemolysis may cause hyperkalemia 24 If there is doubt as to whether the hyperkalemia is artifactual or real, one should obtain a lithium heparin–anticoagulated blood sample and promptly harvest the plasma.21
The list of drugs that may cause hyperkalemia is extensive.10 With the exception of overexuberant infusion of potassium chloride in intravenous fluids and administration of drugs that inhibit production or activity of aldosterone (e.g., enalapril, spironolactone), significant hyperkalemia caused by drug administration is rare and generally only occurs if there is underlying renal or adrenal dysfunction. Only the more common causes are provided in Box 16-3.
Decreased urinary potassium excretion may be due to severe primary renal dysfunction (e.g., typically anuric or oliguric primary renal failure), postrenal causes (e.g., uroperitoneum, uretheral obstruction) or to secondary renal dysfunction resulting from aldosterone deficiency (i.e., hypoadrenocorticism). Disorders promoting development of edema or cavitary effusions or enteritis (especially whipworm infection)25–27 are sometimes associated with the development of mild hyperkalemia, the exact pathogenetic mechanism of which remains unclear. The clinician should rule in or rule out hypoadrenocorticism early in the diagnostic evaluation because it has an excellent prognosis if recognized and treated promptly. An adrenocorticotropic hormone (ACTH)–stimulation test is required for definitive diagnosis, although resting serum cortisol concentrations may be used to screen for its presence. Dogs with resting cortisol levels of more than 2 ug/dL rarely have hypoadrenocorticism,28 and in such cases the submission of a post-ACTH (stimulated) cortisol sample may not be indicated.
Hypokalemia
Increased loss of potassium is the most important cause of hypokalemia in dogs and cats. Vomiting and diarrhea lead to gastrointestinal losses, whereas drug therapy (e.g., furosemide) and renal failure or polyuria result in renal losses. The latter is commonly observed in older cats and may be detected in the absence of azotemia. Hyperaldosteronism is an uncommon disorder of cats that may cause profound hypokalemia. If the cause of hypokalemia is unclear after routine evaluations, the clinician may calculate the fractional excretion of potassium (FEK). Animals that are hypokalemic as a result of nonrenal causes should have normal (approximately 4% to 6%) to decreased FEK, whereas cats with potassium-losing nephropathies usually have values above 6% to 10%.8,29,30
Miscellaneous Minerals
Miscellaneous minerals that occasionally are of concern to the clinician are phosphorus and magnesium. Severe hypophosphatemia capable of causing clinical signs is principally seen in ketoacidotic diabetic patients that are overtreated with insulin and occasionally in emaciated cats receiving enteral or parenteral nutrition,31,32 although hypophosphatemia can be seen from time to time in patients with other diseases.33 The most clinically relevant consequence of hypophosphatemia is hemolysis. Hyperphosphatemia is commonly seen in patients with renal disease but seldom requires special considerations when formulating fluid therapy. Magnesium is primarily absorbed in the jejunum and ileum, while the kidneys regulate magnesium balance. Hypomagnesemia is primarily due to renal or gastrointestinal losses.34 Hypomagnesemia may cause cardiac dysrhythmias and neuromuscular irritability. Hypomagnesemia is increasingly recognized feature of ketoacidotic diabetes mellitus. It is also seen in dogs with severe protein-losing enteropathy.35 Depletion of magnesium has a permissive effect on potassium exit from the ICF leading to ECF accumulation of potassium, which is subsequently lost from the body. This potassium deficiency is refractory to supplementation until the magnesium deficit is also corrected. Similarly, hypocalcemia may occur as a secondary electrolyte abnormality when there is a magnesium deficit. Hypermagnesemia is primarily associated with renal disease. Excessive provision of magnesium (e.g., in diet or intravenous fluids) is also reported.36,37
Acid–Base Status
Traditional evaluation of acid–base status is best accomplished by blood gas analysis, integrating information provided by the pH, pCO2, and bicarbonate. When evaluating blood gas values, consideration is first given to pH.11 The normal pH of blood analyzed at 37° C is 7.35-7.45. If the pH is abnormal, is the patient acidemic (pH < 7.35) or alkalemic (pH > 7.45) If it is abnormal, by definition an acid–base disorder exists. If the pH is normal, an acid–base disorder is unlikely but should not be ruled out, because there may be a mixed acid-base disturbance (i.e., more than one primary acid–base disturbance occurring concurrently) or (uncommonly) a fully compensated acid–base disturbance. These circumstances are not always readily distinguishable. The reader is referred to other references for information on mixed disorders.38
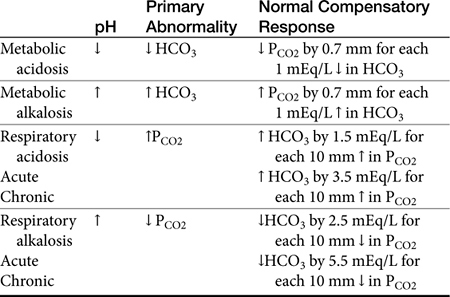
If the pH is abnormal, the clinician should try to determine which component (respiratory or metabolic) is the primary contributor (Table 16-1). Subsequently, evaluation for appropriate compensation of the primary disorder is undertaken. Generally, the pH will vary in the direction of the primary disorder. The other component is the secondary or compensatory component attempting to restore pH to normal. When both components vary in the same direction at the pH, both disorders are primary, i.e., a mixed disorder is present. In the dog there are guidelines for expected compensation in acid-base disorders (see Table 16-1); however, it is not possible to extrapolate these guidelines from the dog to the cat. Results of blood gas analysis are interpreted in light of anamnesis and clinical examination findings to most appropriately determine the most probable cause of the acid–base disorder.11
Metabolic Acidosis
Metabolic acidosis is probably the most widely recognized canine and feline acid–base disorder. The most common causes of metabolic acidosis in these species are listed in Box 16-4. Acidosis from accumulation of lactic acid is thought to be the most common cause of metabolic acidosis. In the setting of poor peripheral perfusion, anaerobic metabolism often predominates, with subsequent production of lactic acid.39 The acidemia is not actually due to dissociation of the lactate; the lactate simply reflects the anaerobic metabolism that has released protons. Therefore lactic acidosis may be seen in conjunction with other disorders. Lactic acidosis is usually diagnosed by elimination of other causes of acidosis, presence of elevated anion gap, and physical examination findings suggesting ischemia or volume depletion. Blood lactate levels may be measured (i.e., hand-held monitors or standard blood gas analyzers).20,21,40 The anion gap, although reported with other serum chemistry measurements, is actually a calculated value [(Na + K) – (HCO3 + Cl)]. The electrolytes used in the formula are called measured cations (Na, K) and measured anions (HCO3, Cl). Ions not included in the formula are called unmeasured cations and unmeasured anions.
Most dogs and cats with diarrhea do not develop a significant acidosis. Occasionally, patients with profuse diarrhea lose excessive amounts of bicarbonate in the feces and consequently develop an inorganic (hyperchloremic) metabolic acidosis. This acidosis may occur simultaneously with lactic acidosis in some patients with concomitant dehydration or hypovolemia.20
Renal failure causes acidosis when the kidneys cannot adequately excrete H+, regenerate HCO3−, or both. Renal ammonium excretion is the principle means of eliminating protons, and this is usually adequate until renal failure becomes severe. Acidosis from renal dysfunction may have a normal anion gap initially, but an increased anion gap is expected in more severely uremic patients as unmeasured anions accumulate. Hypoadrenocorticism causes an acidosis in part because aldosterone is needed for the secretion of H+ into collecting duct fluid; thus it may be thought of as a functional renal disease (i.e., without structural nephron damage). Specific renal tubular defects (renal tubular acidosis) are uncommon in dogs and cats but should be on the list of differential diagnoses for patients with a hyperchloremic acidosis and normal anion gap.20
Diabetic ketoacidosis occurs when there is excessive production of ketone bodies, especially b-hydroxybutyrate, generally causing an increased anion gap acidosis.41
When ethylene glycol is ingested, it is metabolized by the liver to glycolic acid. This often results in severe acidemia that is refractory to standard treatments, largely because acid metabolites continue to be produced until all of the ethylene glycol is metabolized.42 The acidosis typically occurs before there is any evidence of renal injury. History may be informative, and several quantitative colorimetric in-house test kits are available. Calcium oxalate crystalluria often precedes overt renal failure. Both monohydrate (i.e., “picket fence”) and dihydrate (i.e., “Maltese cross”) calcium oxalate crystals may be observed in urine sediment. Markedly increased anion and osmolol gaps are seen shortly after exposure.43 A high anion gap acidosis may be documented within 3 hours and persists for at least 24 hours.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


