Fig. 4.1
Factors affecting evolution of influenza viruses
4.1 Evolution of Haemagglutinin Gene of Influenza Viruses
Compared to influenza C viruses, influenza B viruses are more closely related to influenza A viruses on the basis of detailed molecular studies of NP and polymerase subunits (Gammelin et al. 1990; Krossoy et al. 1999; Cox et al. 2000). Multifunctional haemagglutinin (HA) is the major envelope glycoprotein of influenza A and B viruses, whereas the equivalent protein in influenza C viruses is called hemagglutinin-esterase (HE). Based on their antigenic properties, 18 different HA subtypes (H1–H18) genes of Influenza A virus have been described (WHO Memorandum 1980; Tong et al. 2012, 2013). The sequence data has been used to classify various HAs found in influenza A viruses into two groups. H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17 and H18 are included in group 1, while H3, H4, H7, H10, H14 and H15 form group 2 (Tong et al. 2012, 2013). Aquatic birds are the natural reservoirs of all the HA subtypes except H17 and H18. The introduction of new subtypes of HA genomic segments from aquatic birds is generally responsible for Influenza A virus pandemics in humans. The divergence and alteration in HA genes of the influenza viruses occurs much in advance compared to the other genomic segments in different influenza A virus subtypes. The substitution of amino acids at the rate of 3.19 × 10−4 per site per year for influenza A virus haemmagglutinins from duck was reported, which was found to be similar to that of influenza B and C virus HAs (HEs), though it occurred much slower than that for haemmagglutinins of human and swine A viruses. It might have taken several thousand to several hundred years for these divergences between different subtypes of A virus HA genes to occur (Suzuki and Nei 2002; Nei and Kumar 2000; Nei et al. 2001). The violation of the molecular clock can be used to get a rough estimate of divergence times. It can be hypothesised that evolution rate of amino acid substitution of 10−4 per site per year influenza virus HA (HE) genes in the natural reservoir several thousand of years ago initially led to divergence of these genes into influenza A, B and C virus HA (HE) genes and subsequently into various subtypes in influenza A viruses. Phylogenetic analyses may sometimes lead to overestimation of mutation rates in HA of influenza viruses (Radomski et al. 2014).
Phylogenetic analysis and maps have been used to determine movement and journey of past viruses, their relationship with the current ones and also for identifying a ‘common ancestor’ of the virus. The sequence of the HA gene of the Spanish influenza virus that caused the 1918 pandemic was closer to avian strains than other mammalian ones (Reid et al. 1999; Anhlan et al. 2011). The analysis of the phylogenetic studies conducted on the influenza viruses revealed that the common ancestor of influenza virus goes back prior to the 1918 outbreak that associates the current human virus to the swine virus (Gorman et al. 1990), whereas, the ancestor is derived from an avian host. Therefore, the probability of a virus strain having a large number of mutations to become the generator of a new lineage in future influenza seasons increases (Fitch et al. 2000). The adaptation of HA for virus replication within host is not only influenced by HA sequence and structure but also by the host selection pressures affecting its binding (Wilks et al. 2012). The affinity and avidity of HA influenza A (H3N2) virus for the human and avian HA receptors have changed during the last 45 years. The altered and decreased binding of these recent H3N2 isolates has been found to be due to marked differences in the conformation of the 220-loop of HA1 relative to the 1968 structure, as well as to D225N substitution in the HA1 (Lin et al. 2013). The mode of evolution of HA and NA genomic segments of H3N2 influenza viruses has been found to be quite different (Sandie and Aris-Brosou 2014).
4.2 Role of Receptor Specificity in the Evolution of Influenza Viruses
The influenza A viruses initiate the infection process by recognising specific receptors through HA. The receptors recognised by influenza viruses present in different host species are given in Table 4.1.
Table 4.1
Influenza virus receptors in various host species
Host species | Receptor(s) |
|---|---|
Waterfowl/ducks | Neuraminic acid α-2,3 sialic acid |
Chicken (most chicken isolates except H9N2) | Neuraminic acid α-2,3 sialic acid |
Chicken (H9N2 chicken isolates) | Neuraminic acid α-2,6 sialic acid |
Human | Neuraminic acid α-2,6 sialic acid |
Sea mammals | Neuraminic acid α-2,6 sialic acid |
Pigs | Neuraminic acid α-2,3 and α-2,6 sialic acid |
Horses | Neuraminic acid α-2,3 sialic acid |
Dogs | Neuraminic acid α-2,3 sialic acid |
The HA receptor specificity can be altered through certain mutations in HA gene as well as by host cell selection (Couceiro et al. 1993; Rogers and Paulson 1983; Rogers et al. 1983; Horimoto and Kawaoka 1994; Horimoto and Kawaoka 1994; Stevens et al. 2006; Chutinimitkul et al. 2010; Liu et al. 2010; Xu et al. 2010; Abed et al. 2011; Belser et al. 2011; Wilks et al. 2012; Lin et al. 2012; de Vries et al. 2014). The HA present on human influenza virus preferentially binds to cell receptors containing α-2,6 sialic acid residues, whereas avian influenza virus (AIV) preferentially binds to α-2,3 sialic acid residues (To et al. 2012). Human tracheal epithelial cells have mainly α-2,6 linked residues (Rogers and D’Souza 1989; Conner et al. 1994; Olofsson et al. 2005; Imai and Kawaoka 2012). Swine respiratory epithelium has a mixture of receptors with α-2,3 and α-2,6 linkages (Ito et al. 1998). Studies using various mutants revealed that differences in both the oligosaccharide chains and the amino acid sequences around the receptor binding site of HA can influence the receptor recognition, and receptor binding is important for replication and production of progeny of avian viruses in humans (Chen et al. 2006, 2012). The α-2,3 specific highly pathogenic AIVs (H5N1) must acquire α-2,6 sialoside receptor specificity for efficient transmission in humans. A reassortant human avian influenza virus exchanging gene segments of HA gene can possibly change the receptor specificity of the virus from avian type receptor (α-2,3 sialic acid) to human type receptor (α-2,6 sialic acid). The human infections due to subtype H5N1 in Hong Kong in 1997 was speculated to be caused by such a human reassortant containing avian HA gene (Olofsson et al. 2005). The changes in the receptor specificity of the avian virus HA can on some occasions result in interspecies transmission of AIVs circulating in wild aquatic birds to mammalian animals and human, causing influenza in them (Matrosovich et al. 2000, 2001, 2009; Abdel-Moneim et al. 2012; Watanabe et al. 2012). Besides the host cell surface receptor, the avian- and human-adapted influenza viruses will also require a cellular protein, importin-α isoform, for transport of the viral ribonucleoprotein complex into host cell nucleus (Gabriel et al. 2011). Differences have been reported in the expression patterns of influenza virus receptors in various organs in chickens, ducks, pheasants and quails (Kuchipudi et al. 2009; Yu et al. 2011) which may have implications in the interspecies transmission of influenza viruses.
4.3 Role of Antigenic Drift and Antigenic Shift in the Evolution of Influenza Viruses
Influenza viruses are dynamic and are continuously evolving by two different mechanisms, viz. antigenic drift and antigenic shift (Fig. 4.2) (Wikramaratna et al. 2013; Stower 2014). Antigenic drift occurs due to errors during replication, which are irreparable. The change produced by antigenic drift in three influenza virus types (Influenza A, B and C) is occurring all the time because of its error-prone polymerase resulting in accumulation of genetic mutations that are selected for HA and to a lesser extent neuraminidase (NA), i.e. the major surface glycoproteins of the virus (Hensley et al. 2009; Lambert and Fauci 2010). The antibodies—or immune responses—induced selection pressure is responsible for such point mutations (Scholtissek 1995). Antigenic drift refers to small, gradual changes that occur through point mutations in the genes for surface proteins, haemmagglutinin and neuraminidase. The mutation frequency is <1 % per year in the amino acid sequence of human HA and NA. Five antigenic sites in the HA of influenza A viruses (designated as A, B, C, D, E for the H3 subtype; and Ca11, Ca2, Cb, Sa, Sb for H1 subtype) have been identified (Enami and Enami 1996; Ruigrok et al. 2000; Sanz-Ezquerro et al. 1995). Antigenic variation can occur even with a single point mutation in one HA antigenic site (Salvetore et al. 2002; Sanz-Ezquerro et al. 1995; Scheiffele et al. 1997; Tombari et al. 2013). Similarly, amino acid differences have been reported to be responsible for the antigenic drift in the neuraminidase (Burleigh et al. 2005; Luo et al. 1991). However, antigenic shift happens only occasionally in viruses with segmented genomes. The latter term denotes the exchange of one or more genome segments between two related viruses which infect a host cell at the same time. During such a double infection, various components of both viruses are generated during replication of these viruses in the same host cell. During the subsequent assembling of the virus, mistakes in the combination of the segments can happen because the system cannot differentiate which RNA segments emerge from which subtypes. The reassortment of complete units of genetic material results in the formation of ‘mosaic’ viruses that are known as ‘reassortants’. At times, the antigenic shift/genetic reassortment affects the exchange of genome segments encoding the viral surface proteins HA and neuraminidase. Thus, the virus gains a new antigenic pattern/specificity, the process so-called ‘antigenic shift’. While this is a common phenomenon, it is also the catalyst for pandemics among humans because pandemic viruses are often genetic reassortants of human and avian/swine influenza A virus subtypes. Although avian influenza A subtypes usually cannot infect humans and poultry is usually not susceptible to human virus subtypes, pigs play an important role in the formation of new influenza viruses. They serve as a kind of ‘melting pot’ as they are susceptible to double infections with avian as well as human influenza viruses. In this way, new virus variants can be transmitted from pigs to humans. The risk of jumping the species barrier between birds and humans is particularly high for the lethal avian flu virus H5N1, even though it caused only few infections of humans until now. Formation of new viral subtypes is a remarkable example for the effects of antigenic shift. Besides these obvious changes, less palpable reassortments and mutations of the segments happen, which are accountable for the formation of different genotypes within one subtype. Consequently, 9 different genotypes of the avian flu virus H5N1 have been reported so far (Li et al. 2004), which differ in their degree of pathogenicity.
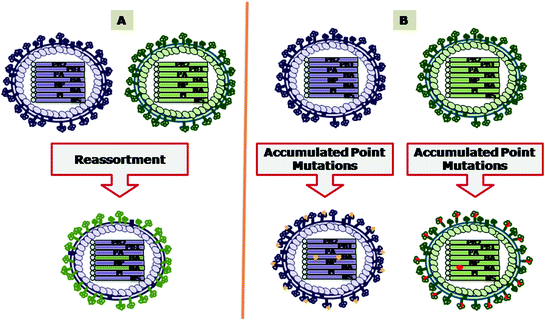
Fig. 4.2
Evolution of influenza A virus by A antigenic shift and B antigenic drift. The exchange of the genomic segments through the process of reassortment is responsible for antigenic shift. The accumulation of point mutations results in antigenic drift
The mortality caused by H5N2 isolated from the chickens in Pennsylvania had increased from <10 % in April 1983 to >80 % in October 1983. Both the April and October H5N2 isolates had multiple basic amino acids in the HA cleavage site. However, there were seven nucleotide differences resulting in four deduced amino acid changes. The loss of the glycosylation site by one such change exposed the multiple basic amino acids, thus, facilitating the cleavage and activation of HA by furin and PC6, the ubiquitous cellular proteases (Holsinger et al. 1994). There are also many more instances where viruses of low pathogenicity, after circulation for some time in a poultry population, mutated into highly pathogenic virus, e.g. H5N2 in 1994 in Mexico, H7N3 in 1995 in Pakistan, and H7N1 in 1999–2001 in Italy, H7N3 in 2012 in Mexico (Horimoto et al. 1995; Li et al. 2001; Panigrahy et al. 2002; Goot et al. 2003; Hall 2004; Wainwrighta et al. 2012). Normally, the waterfowl flu viruses are avirulent in land-based poultry. However, sometimes point mutations may occur in these viruses during their replication in land-based poultry. This may result in NA stalk deletions and acquisition of additional glycosylation sites. The adaptation of AIVs from waterfowl to domestic poultry due to NA stalk deletion occurs by selection and not by de novo emergence of viral mutants (Croville et al. 2012). These viruses now have low affinity for SA α-2,3 Gal and, thus, resemble the human viruses (Matrosovich et al. 1999, 2001). It has been observed that adaptive mutations that result in enhanced polymerase activity can contribute to high virulence of influenza A virus in mice (Rolling et al. 2009). Antigenic drift can also affect the susceptibility of influenza A viruses to antiviral drugs. The amino acid substitution at 274th position of the NA and H275Y mutation in NA protein sequence of some H5N1 viruses has been implicated in their resistance to oseltamivir and other neuraminidase inhibitor drugs (de Jong et al. 2005; Anton et al. 2012). The virus lineage determines the outcome of E627 K mutations in PB2 segment of HPAI H5N1 (Long et al. 2013). However, the same mutation is responsible for the high polymerase activity and enhanced replication of H7N9 influenza virus (Zhang et al. 2014). The mutations at positions 182 and/or 192 in the HAs of H5N1 viruses will enable the mutated viruses to recognise the human receptor instead of the avian receptor. These amino acid changes can be used as molecular markers for determining potential of H5N1 field isolates to cause pandemics (Yamada et al. 2006). There are 11 neuraminidase types which have further been reported to diverge into 23 NA lineages (Xu et al. 2012).
Antigenic drift produces new virus strains that may not be neutralised by antibodies to earlier influenza strains. It produces an increasing array of strains until one evolves that can infect human/animals who have protective immunity to the pre-existing strains. The variants produced by antigenic drift can sometimes cause epidemics and usually prevail for 2–5 years before being replaced by a new variant. The infection of a person with a specific influenza virus strain leads to the development of antibodies against that strain in due course of time. The infection with a newly emerged strain can occur if the antibodies against the older strains are unable to recognise the newer virus. It is for this reason that people can become infected with influenza viruses many times. Therefore, global surveillance is essential for monitoring the evolution of human influenza virus stains for selection of appropriate strains that should be included in the annual influenza virus vaccine production. Normally, one or two virus strains in the influenza vaccine are updated every year to keep abreast with the changes in the circulating influenza viruses. Therefore, annual vaccination against influenza is needed for protection.
Antigenic shift refers to an abrupt, major change to produce novel influenza virus subtypes that was not currently circulating or prevailing among population. Antigenic shift can take place either through direct animal (poultry/swine)-to-human transmission or through mixing of human influenza A and animal influenza A virus genes through a process called genetic reassortment in so-called ‘mixing vessels’ (swine/quails) or in reservoir hosts including migratory/free living birds and large populations of domestic poultry when co-infected with more than one influenza virus strain (Swayne and Halvorson 2003). The influenza viruses are capable of reassortment of segments from other influenza virus strains within a genus or type during mixed infection. However, reassortment among the different genera has not been observed. The genome of influenza A or B viruses have eight segments. Thus, the mixed infection with two strains within influenza A or B viruses can have the possibility of generation of 28 influenza A viruses (2 parent strains and 254 genetically different progeny viruses). Similarly, theoretically 128 different influenza C viruses (which include 2 parent strains) can be generated during mixed infection with influenza C viruses.
Antigenic shift due to reassortment plays an important role in the evolution of influenza A viruses and its consequences. The reassortment of two AIV strains under natural conditions and subsequent infection of seals with this reassortant caused heavy mortality in 1979 (Scholtissek 1995). Reassortant virus, H2N2, which acquired the HA, NA and PB1 genes from Euroasian AIVs in a human, H1N1, genetic background was responsible for the 1957 ‘Asian Flu’ pandemic. Similarly, the 1968 ‘Hong Kong Flu’ human pandemic was caused by avian–human reassortant (Reid et al. 1999; Horimoto and Kawaoka 2001), H3N2, which evolved due to replacement of HA and PB1 segments on human H2N2 virus background with genomic segments from avian origin (Kawaoka et al. 1989; Scholtissek et al. 1978). The severity and mortality in the 1968 pandemic was less than the 1957 pandemic. The reason for this is reported to be the presence of pre-existing antibodies to N2 protein which was common in both the reassortant viruses that caused these two pandemics. The reassortment between H1N1 swine viruses and human H3N2 viruses generated an H1N2 reassortant virus in Japan in 1978, and which has since established throughout the Japanese swine population (Sugimura et al. 1980). The reassortment may not be limited between two strains as many reassortants containing genes derived from human, swine and avian influenza A viruses have been identified. Many of the swine influenza viruses isolated from pigs in the United States since 1998 were found to be triple reassortants as the genome of these viruses contained genomic segments derived from swine, avian and human influenza A viruses (Karasin et al. 2000; Webby et al. 2000; Zhou et al. 1999; Olsen et al. 2002, Trifonov et al. 2009; WHO 2009). In this triple reassortant, only NP, M and NS gene segments came from swine virus origin; while PB2 and PA genes were from avian origin; the remaining HA, NA and PB1 were contributed by the human influenza virus. The swine reassortant viruses have been shown to infect humans (Olsen et al. 2006; Gray et al. 2007; Newman et al. 2008; Gray and Kayali 2009; Shinde et al. 2009). The swine-origin influenza A (H1N1) strain (S-OIV) (commonly called as swine flu and which was responsible for the latest human pandemic), is a new reassortant containing six gene segments from the known triple reassortant swine virus, and two gene segments (NA and matrix protein) from the Eurasian influenza A (H1N1) swine virus lineage (Solovyov et al. 2009; Christman et al. 2011). Multiple level reassortments were involved in the emergence of this final reassortant (Fig. 4.3). Initially, a reassortment between two influenza A (H1N1) swine viruses occurred. These two viruses themselves were generated due to at least four independent avian-to-mammalian cross-species transmissions, with at least four previous reassortments of gene segments among avian, human and swine-adapted viruses (Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team 2009; Garten et al. 2009; Smith et al. 2009; Zimmer and Burke 2009). The efficiency of human-to-human spread of most of the triple reassortant swine influenza A (H1) viruses found in pigs is low, although these may occasionally be transmitted to humans. The S-OIV, in contrast, has shown human-to-human transmission in many countries (Belshe 2009). Also, the S-OIV is not able to cause any epidemic in pigs, although infection of the pigs may occur due to their exposure to infected humans. Identification of the origin of viral genes in a triple reassortant pandemic 2009 influenza strain by high resolution mass spectrometry proteotyping was found to be better than RT-PCR assay (Fernandes and Downard 2014).
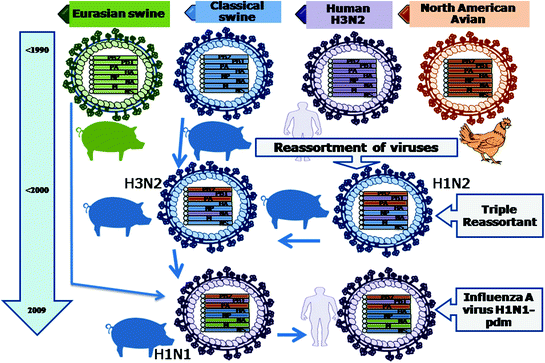
Fig. 4.3
Evolution of influenza A virus H1N1-pdm. Reassortment among classical swine influenza virus, human H3N2 influenza virus and North American avian influenza virus in pigs led to the formation of a triple reassortant H1N2 having PB2 and PA genomic segments from North American avian influenza virus, PB1, HA and NA genomic segments from human H3N2 influenza virus, M, NP and NS genomic segments from classical swine influenza virus. This triple reassortant underwent reassortment with classical swine influenza virus in pigs to produce H3N2 reassortant. This H3N2 reassortant further reassorted with Eurasian swine influenza virus in pigs in the year 2009 to produce a H1N1 influenza virus. This H1N1 virus after replication and adaptation in pigs infected the human and evolved as Influenza A Virus H1N1-pdm
Multiple reassortment processes among AIVs led to the generation of the H5N1 viruses (Fig. 4.4), which affected and caused mortality in humans and chickens. Although all the genomic segments of this virus are of avian origin (Class et al. 1998; Li et al. 2004; Subbarao et al. 1998), nevertheless, it was a reassortant virus. Its six genes (PB2, PB1, PA, NP, M and NS) came from an avian virus (Guan et al. 1999) while NA gene originated from another avian N1 virus and the HA gene was acquired from A/goose/Guangdong/1/96 (H5N1)-like viruses (Xu et al. 1999). Virus genotypes with multiple reassortments were detected in studies conducted on avian H7 influenza viruses circulating in Eurasia during 1999–2005 (Campitelli et al. 2008). The huge genetic diversity observed among the H9N2 and H5N1 influenza viruses in southern China occurred as a result of reassortment among multiple lineages of H5N1 and H9N2 viruses co-circulating for a long period of time in diverse types of poultry. Various types of poultry have been found to display two-way interspecies transmission of influenza viruses. This situation may favour the emergence of influenza viruses with potential to cause pandemics in humans (Xu et al. 2007a, b). The sequence analysis of avian/swine virus reassortant H2N3 influenza A viruses isolated and characterised from diseased swine revealed the presence of leucine at position 226 of the H2 protein. The increased binding affinity of this reassortant to the mammalian 2, 6 Gal-linked sialic acid virus receptor is attributed to this amino acid change (Ma et al. 2007). An unusual reassortant of swine influenza H1N2 virus in Germany has been reported (Zell et al. 2008a, b). The transmission of reassortant swine influenza H3N2 virus to turkeys in USA has been reported (Choi et al. 2004). The complete-genome approach is required to detect new influenza strains and future epidemics as reassortment among various lineages may continue to occur (Holmes et al. 2005; Simonsen et al. 2007; Vana and Westover 2008). Swine influenza viruses containing one or more segments of human seasonal influenza virus origin have been detected (Cappuccio et al. 2011; Ducatez et al. 2011; Moreno et al. 2012). Recently, various hotspots for reassortment among influenza virus have been predicted (Fuller et al. 2013).
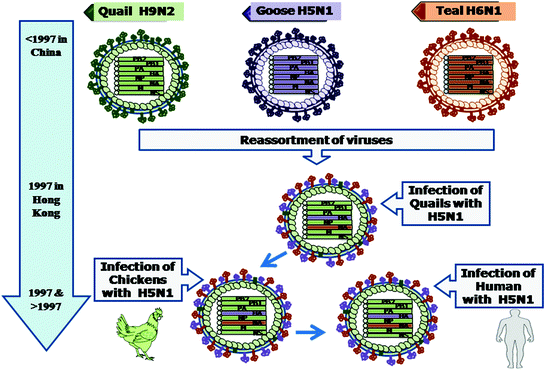
Fig. 4.4
Evolution of influenza A virus H5N1. Reassortment among Quail H9N2, Goose H5N1 and Teal H6N1 in quail led to the formation of a reassortant having HA genomic segment from goose, neuraminidase from teal against the background of the six other genomic segments coming from quail (H9N2) influenza virus. This chicken-adapted H5N1 influenza virus evolved as H5N1 influenza virus that was able to infect human from 1997 onwards
4.4 Role of ‘Mixing Vessels’ in the Evolution of Influenza Viruses
The high population density of human beings, trend of backyard poultry (ducks, geese and chickens) and piggery gives ample chances for intricate and close interaction between these influenza reservoir animals and enhances the probability to a great extent for the interspecies transmission and genetic reassortment. The influenza virus cannot spread freely from any host species to any other species. There are certain limitations, e.g. direct pathway between humans and birds are known to exist. Pigs, however, serve as an open pathway. Therefore, pigs are considered as donator of the virus relatively easily and can act as ‘mixing vessel’ or ‘mixing bowl’ for genetic reassortment and are known to be permissive for both the human (Chambers et al. 1991) and avian influenza (Hinshaw et al. 1981; Schultz et al. 1991) viruses. Pigs have, therefore, been considered and implicated as an intermediate host for the origin influenza viruses causing pandemics (Scholtissek 1990; Ito et al. 1998; Brown 2000; Ito 2000; Peiris et al. 2001; Permin 2004; Ma et al. 2007; Nava et al. 2009; Ma et al. 2009; Kuntz-Simon and Madec 2009; Zell et al. 2013). This phenomenon is due to the presence of both α-2,3 and α-2, 6-galactose sialic acid linkages in cells lining the respiratory epithelium of pig’s trachea which can modify the receptor binding specificities of AIVs from α-2,3 to α-2, 6 linkage (Ito et al. 1998). The α-2,6 linkage being the natural linkage in humans, grants a potential link from birds to humans (Brown 2000). This could result in the emergence of a ‘new AIV’ possessing essential genes from human influenza virus that permits its replication and spread in the human population causing pandemics of devastating potential as the immune system of the human beings would be naïve to the reassortant virus. The outbreak of avian-to-human direct transmission of influenza A viruses that occurred in Hong Kong during 1997 supports the role of pigs as a source of potentially dangerous influenza A viruses (Ito 2000). In China, there are reports of co-circulation of avian H9N2 and human H3N2 viruses in pigs, which might create an environment for genetic reassortment to occur between them, and subsequently resulting in the emergence of viruses with pandemic potential (Peiris et al. 2001). Studies have reflected that quails can also act as an intermediate host and as an ‘avian mixing vessel’, and may have played a significant role of route modulator in evolution of influenza viruses (Makarova et al. 2003; Perez et al. 2003a; Shortridge et al. 2003; Ebrahimi et al. 2011; Thontiravong et al. 2012). Quail have been shown to support the replication of at least 14 AIV subtypes (Makarova et al. 2003; Yamada et al. 2012). Studies have also indicated that quails could provide an environment in which viruses like the swine H3N2 subtype could further reassort and generate new viruses (Perez et al. 2003a, b; Makarova et al. 2003; Thontiravong et al. 2012). It was demonstrated that a human avian recombinant influenza virus, possessing the surface glycoprotein genes of a quail virus and the internal genes of human influenza virus, replicated and got transmitted easily in quail. It indicated the probable role of quails as amplifiers of influenza virus reassortants, which may have the capability to infect humans and could generate incipient pandemic situation (Makarova et al. 2003). The mixing vessels lead to the evolution of quasi-species, which are diverse and new viral populations, for which the poultry and human beings are naive. The chronological evolutionary history of pandemic flu viruses can be determined by understanding the role of the ‘mixing vessel’, and various studies on the evolution of 1918 pandemic flu virus has categorically demonstrated the role of pigs as a mixing vessel (dos Reis et al. 2009). Although, the ‘mixing vessel’ concept is now becoming narrower in comparison to previous years, the emergence of human pandemic influenza virus harbouring genes supposed to be of swine origin emphasised on importance of epidemiology of influenza virus in pigs (Smith et al. 2009).
4.5 Role of Recombination in the Evolution of Influenza Viruses
The phenomena of recombination, particularly by template switching, can also occur in influenza viruses, and thus, provide another mechanism for the evolution of influenza viruses (Hao 2011). This may have an effect on the biological properties especially when recombination involves HA gene. The cleavability of HA was enhanced by 60-nucleotide insertion from NA gene into A/Seal/Massachusetts/1/80 virus (Orlich et al. 1994); and by 54-nucleotide insertion of 28S rRNA into A/Turkey/Oregon/71 (Khatchikian et al. 1989). The insertion of 30 nucleotides of the NP segment (Suarez et al. 2004) or 21 nucleotide of the matrix segment (Bowes et al. 2004; Hirst et al. 2004; Pasik et al. 2005) into the HA segment was responsible for increased pathogenicity of AIVs from Chile and Canada, respectively. The insertions of parts of PB1, PB2 or NP genomic segments into the NA stalk increased the efficiency of replication of A/WSN/33 in embryonated eggs (Suarez et al. 2004). Additional glycosylation sites on HA and amino acid deletion at stalk region of neuraminidase might also change the antigenic nature of the virus, and inhibit some of the host’s antibodies from effectively neutralising the virus (Baigent and McCauley 2001).
4.6 Role of ‘Host Species Jumping’ in the Evolution of Influenza Viruses
In addition to point mutations, reassortment and recombination, new influenza viruses in mammals can evolve by host species jumping (Neumann and Kawaoka 2006; Yamada et al. 2010; Murcia et al. 2012; Reperant et al. 2012; Rahnama and Aris-Brosou 2013; Resa-Infante and Gabriel 2013; Steinhauer 2013; Zhang et al. 2013). The alteration, modifications and changes in the receptor specificity of influenza permit AIVs to overcome the species barrier. The direct transmission and infection of avian H5N1 and H9N2 influenza viruses from chickens and/or quails to humans signifies that the pigs may not always be required to act as an intermediate host for AIV infection of humans. Quails and chickens themselves have the capacity to act as intermediate hosts for transmission of AIV to humans (Thontiravong et al. 2012). It was thought that the human influenza virus is retained in humans only and does not spread to other species (Liu et al. 2009). However, the transmission of pandemic H1N1 and some other H1 and H3 influenza viruses from human to pigs has also been reported since 1990 from many different places. Thus, it has given the chance for reassortment due to increased genetic diversity (Nelson et al. 2012a, b).
Host species barriers to influenza virus infections have been reported (Suzuki 2005; Kuiken et al. 2006; Giannecchini et al. 2006; Guo et al. 2007; Musa et al. 2009). The H1N1 influenza virus having all the segments of avian origin caused influenza in European swine in 1979. Similarly, the equine H3N8 influenza virus which affected horses in 1989 in northern China was found to be enormously similar to an avian H3N8 influenza virus, but exceedingly different from the H3N8 equine influenza virus presently prevailing in other geographical locations (Webster and Guo 1991). The first avian isolate of H5N1 isolated in 1959 is distinct from the ones that are presently circulating. None of its successors were able to cross the species barrier. However, the currently circulating H5N1 subtypes have not only crossed/jumped the species barrier to infect animals and humans, but also caused heavy mortality in birds (Suarez 2000; Swayne and Suarez 2000; Capua and Alexander 2002; Beigel et al. 2005; Yuen and Wong 2005; Perdue and Swayne 2005; OIE 2005; WHO 2005; Zeitlin and Maslow 2005; Musa et al. 2009). Domestic ducks have also been implicated in the perpetuation, proliferation and evolution of highly pathogenic H5N1 influenza viruses in Asia (Hulse et al. 2005; Ramirez et al. 2005; Musa et al. 2009). This virus can affect pigs (Permin 2004) and even carnivores such as tigers and leopards (Enserink and Kaiser 2004; Thanawongnuwech et al. 2005; Keawcharoen et al. 2004; Thornley 2004). The avian H5N1 influenza virus has been isolated from dogs (Songserm et al. 2006) and cats (Kuiken et al. 2004; Songserm et al. 2006; Thiry et al. 2007; Desvaux et al. 2009). The infection and disease is produced in humans by other AIVs such as H9N2 in China and Hong Kong in 1998–1999 (Guo et al. 1999; Peiris et al. 1999); H7N7 in Netherlands in 2003 (Fouchier et al. 2004); and H7N3 in 2004 in Canada (Tweed et al. 2004); H7N5 and H10N6 (To et al. 2012) has also been reported. The avian H9N2 viruses have been detected isolated from wild aquatic birds in Argentina (Xu et al. 2012) and pigs in Southeast Asia (Peiris et al. 2001), and isolation of H5N1 avian viruses have been reported from Thailand and Vietnam (Choi et al. 2004). The equine H3N8 has been isolated from racing greyhounds suffering from respiratory disease (Crawford et al. 2005; Crispe et al. 2011). The severe respiratory disease in English foxhounds was also found to be caused by an equine influenza A virus (H3N8). The canine respiratory tissue was shown to possess the relevant receptors for infection with equine influenza virus (Daly et al. 2008). Direct transmission of AIV (H3N2) from poultry to dogs is possible due to the observation of sal α-2,3-gal receptor in parts of the respiratory system of dogs. The isolation of avian H3N2 from dogs provided evidence that dogs may play a role in interspecies transmission and spread of influenza virus (Song et al. 2008; Mancini et al. 2012). The avian H10N4 virus caused death in minks in Sweden (Klingeborn et al. 1985). The viral pneumonia in seals in the coastal regions in USA was found to be caused by avian H7N7 in 1979 (Webster et al. 1981), and by avian H4N5 in 1982–1983 (Hinshaw et al. 1984). The AIV (H3N8) was recently found to infect harbour seals in New England, USA (Anthony et al. 2012).
Various viral and host factors are involved in the evolution of influenza virus which is a dynamic and continuing process (Webster et al. 1992; Makarova et al. 2003; Webster and Hulse 2004; Permin 2004; Matsui 2005). A convergence of factors, including the combination and interaction of many of these factors such as the population densities of poultry, free living/migratory birds and humans, ecology and physiological traits of hosts (Cobey et al. 2010), weather and temperature conditions can influence the evolution of the influenza viruses (Fuhrmann 2010). Highly dense poultry and pig farming, in concurrence with conventional live animal or ‘wet’ markets also play a role in the genesis of reassortant viruses (To et al. 2012). This congenial environment for the influenza virus provides conditions in which the ‘mixing vessels’ get the opportunity to generate new viruses by increased mutation, reassortment and recombination events.
The intra-host and inter- host evolutionary dynamics, and evolutionary basis of cross-species transmission will help in understanding the mechanisms that influence the emergence of either novel viruses or variant antigenic strains capable of causing pandemics (Nelson and Holmes 2007; Rambaut et al. 2008; Russell et al. 2008; Murcia et al. 2010; Taubenberger and Kash 2010). The genomic signatures of influenza A viruses were identified on the basis of adjusted Rand index (ARI) for their ability to distinctly infect various hosts (Hu et al. 2014). Novel avian influenza A H7N9 viruses have been found to infect humans (Lebarbenchon and Stallknecht 2011; Gao et al. 2013; Lebarbenchon et al. 2013; Liu et al. 2013a). These influenza viruses overcame host species barriers by acquiring adaptive mutations in the PB1, PB2, PA, NP, NEP (Mehle et al. 2012; Paterson and Fodor 2012; Manz et al. 2013); NS1 (Noronha et al. 2012) or NP and M genes (Ince et al. 2013).
4.7 Evolution of the Highly Pathogenic H5N1 AIVs: Concept of Virus Clades
The highly pathogenic avian influenza (HPAI) H5N1 virus has been detected and isolated from wild birds or domestic poultry in many Asian, European and African countries since 1996 and has caused sporadic infections in humans and raised pandemic concern (Peiris et al. 2007). The H5N1 virus that was circulating in poultry population located at different geographical areas during the last 17 years has changed a lot. It is now showing remarkable genetic diversity as the result of accumulated point mutations.
The isolation of the (HPAI) H5N1 viruses was regularly and continuously reported by various laboratories around the world. Many laboratories referred these as emerging lineages of HPAI, H5N1 and gave their own nomenclature to these isolates. The use of several provisional names, which are different from each other, added lots of confusion and made the comparative analysis to these circulating and/or emerging lineages HPAI, H5N1 a cumbersome and difficult task. Avian H5N1 viruses have continued to evolve, diversify and spread. The infection of animals and humans by these viruses continues to occur. Except the specific H5 HA gene identified in 1996, the reassortment has replaced most viral genes leading to the generation of many different genotypes in all these isolates. The evolving strains may, therefore, be effectively compared with the stable H5 HA. Detailed phylogenetic analysis was performed by a collective international team, identified by the World Health Organization (WHO), the World Organisation for Animal Health (OIE) and the Food and Agriculture Organization (FAO), on all the H5 HA sequences available in the public domain that have evolved from the original A/goose/Guangdong/1996 H5N1 virus. The evolution of this H5 HA formed the base for the development of a standard ‘clade’ nomenclature system. The following criteria developed by the WHO/OIE/FAO H5N1 Evolution Working Group were used to categorise currently circulating H5N1 viruses into various groups called ‘clades’.
1.
The common clade-defining node in the phylogenetic trees are shared;
2.
A bootstrap value of ≥60 at the clade-defining node (after 1000 neighbour-joining bootstrap replicates) will lead to their clubbing into a monophyletic group; and
3.
Average percentage pairwise nucleotide distances of >1.5 % and <1.5 %, within clades will be considered.
With the help of this unified clade system, the sequence and/or surveillance data coming from different sources can be analysed and interpreted easily and more usefully as different clades may be found in the same geographical area and/or same clades may be isolated from different geographical areas. The geographical reference currently used then becomes meaningless. It will also help in the expansion of the phylogenetic tree in future. This will also lay the foundation to develop a system in which the issues of antigenic variation and reassortment into multiple genotypes are properly addressed.
Based on these criteria, 20 distinct clades of the H5 viruses have been identified since its inception in early 2008 (WHO/OIE/FAO H5N1 Evolution Working Group 2009, 2012), Continuous evolution of the viruses within these clades results in the periodic emergence of new sublineages (potential H5N1 clades). These sublineages (or discrete monophyletic groups) will have to be classified as separate clades on fulfilling all the three specific criteria enumerated above that were used to define the initial clades from which these sublineages emerged. The continuous evolution of the strains within the second order clade may reach a level of genetic diversity at which point it may become essential to split it further into third order clades and so on. The same clade designation criteria apply to first, second and any higher order clade designations. A hierarchical decimal numbering system is used to allocate a number or numerical address to these new clades designated as second, third or fourth order clades, which will link them to their original clade. Between 1996 and 2001, there were four designated clades (0, 3, 5, 9) that were further expanded to 10 clades (0, 3, 4, 7, 5, 6, 8, 9, 1, 2) between 1996 and 2004. 5 second order clades (2.1, 2.2, 2.3, 2.4, 2.5) were incorporated in clade 2 in 2005. Three (2.1.1, 2.1.2 and 2.1.3), and four (2.3.1, 2.3.2 and 2.3.3, 2.3.4, 2.3.5) third order clades were made within the distinct clade 2.1 and 2.3, respectively, between 2005 and 2008 on fulfilling the clade definition. The fourth order clade designation have been assigned, between 2008 and 2011 period, to a new monophyletic clades identified within clade 2.3.2 (named as clade 2.3.2.1); 2.3.4 (2.3.4.1, 2.3.4.2, 2.3.4.3). During the period between 2008 and 2011, second order clades 1.1 were made in clade 1; and 2 second order clades (7.1, 7.2) were made in clade 7. The clade 2.2 was further split into third (2.2.1) and fourth (2.2.1.1) order clades during the period between 2008 and 2011. The H5N1 Evolution Working Group has recommended that emerging sublineages of H5N1 can be classified into a separate clade using the hierarchal decimal numbering system only after the sublineage has met all the three clade definition criteria. However, it has also permitted the use of temporary, provisional alternative names for those sublineages of H5N1 virus that are newly emerging and quite distinct, until these can be incorporated into the clade system using the hierarchal decimal numbering system.
Recent phylogenetic analysis of the neighbour-joining tree, constructed from recent ~3000 HA gene sequences data, generated one or more monophyletic groups with high bootstrap support within every clades of H5N1 which is presently circulating in various populations. In addition, further nucleotide divergence was indicated in several of these groups which had long branch lengths that separated them from the nearest node in the tree. Among the currently circulating clades (1, 2.1.3, 2.2, 2.2.1, 2.3.2, 2.3.4 and 7), the average within-group pairwise nucleotide distances were found to have >1.5 % within-group divergence. Consequently, these groups were split into new order clades. Subsequent to the generation of clade-specific trees, the within- and between-group p-distances were determined from the analysis of monophyletic groups with bootstrap values >60. This led to the identification of 12 new second, third and fourth order H5N1 clades as compared to only one new clade identified with the previous nomenclature update. This may be due to increased time span taken for the nomenclature updating between the current and previous. Another reason could be the availability of growing number of sequences that are available for comparative analysis due to improved surveillance as well as better reporting of the viruses in recent years. The largest numbers of new clade designations were required for, and given to those clades which had already diversified and were reclassified into third order groups in previous analyses. Although new clades of H5N1 have emerged of late, an interesting observation has been the disappearance of many of the previously circulating clades of H5N1, which have not been detected since at least 2008. This may be due to deficiency in surveillance. Another reason could be that many of these clades have been displaced, replaced or supplanted by new clades and became dormant or inactive. Thirteen clades (0, 2.1.1, 2.1.2, 2.3.1, 2.3.3, 2.4, 2.6, 3, 4, 5, 6, 8 and 9) have not been detected since 2008 (WHO/OIE/FAO H5N1 Evolution Working Group 2009, 2012).
The wild birds of Qinghai Lake in China suffered widespread outbreaks by clade 2.2 H5N1 virus. This clade subsequently spread westwards, via long-distance bird migration, to the Middle East and South Asia, Europe and Africa in 2006–2007 and resulted in endemic infection in the poultry populations of some Asian and African countries (Li et al. 2011). However, during the same period clade 2.3.4 was the dominant virus clade in Southeast Asia. The clade 2.3.2 viruses have been repeatedly isolated from wild birds and migrating birds in Hong Kong, Japan, Russia and Mongolia, Europe (Kang et al. 2011; Reid et al. 2011). The clade 2.2 H5N1 viruses were isolated from poultry and humans in South Asian countries (India, Pakistan, Bangladesh, Nepal and Bhutan) between February 2006 and January 2010 (Dubey et al. 2009; Chakrabarti et al. 2009; Tosh et al. 2011a, b). The clade 2.3.2 H5N1 virus was reported for the first time in South Asia in Nepal in February, 2010 (Reid et al. 2011). However, the outbreaks in Indian state of Tripura and adjoining Bangladesh during the same period were caused by viruses belonging to a different clade, i.e. clade 2.2 H5N1 viruses (Tosh et al. 2011b). Recently in 2011, clade 2.3.2 H5N1 viruses have been detected in poultry Indian state of Tripura. The results of the phylogenetic analyses on these 2011-outbreak viruses indicated that they belong to clade 2.3.2.1 but were different from the clade 2.3.2.1 viruses isolated in Nepal (Nagarajan et al. 2012). Antigenic analysis of these 2011-Tripura outbreak isolates confirmed 64–256-fold reduction in Haemagglutination-Inhibition (HI) titres compared with clade 2.2 viruses that were prevalent during 2008 outbreaks in the same region. The emergence, proliferation and spread of clade 2.3.2.1 of HPAI H5N1 virus in the South Asian region indicates that this virus clade may have rooted and entrenched itself in wild birds and land-based poultry, and is spreading to new regions similar to the previous clade 2.2. The detection and recognition of the new clade 2.3.2.1 H5N1 viruses in South Asian countries, that are phylogenetically closely linked to those isolated from Qinghai Lake, China, Korea and Mongolia in 2009 and 2010, present a situation similar to the introduction and establishment of clade 2.2 viruses in this region in 2006–2007 (Choi et al. 2013; Xu et al. 2013a). All efforts should be made to examine whether clades 2.3.2.1 and 2.2 are simultaneously circulating in this region or whether clade 2.2 is being displaced and replaced by clade 2.3.2.1. The risk of evolution of pandemic H5N1 strains, with drastic and devastating consequences, will increase due to simultaneous and continued co-circulation of several H5N1 viruses belonging to various clades and subclades. The threats from these viruses have further risen due to better adaption in the land-based poultry in regions of South Asia which has dense human population (Nagarajan et al. 2012). Moreover, clade 2.3.4.2 viruses have recently been isolated from Myanmar and Bangladesh and show decreased serological relationship with clade 2.3.4 viruses.
4.8 H17N10: A New Subtype of Influenza Viruses of Bat
The repertoire of known subtypes of influenza viruses, and the known range of mammals that can be infected by these viruses has increased with the discovery of new subtype, H17N10, from little yellow-shouldered bats caught at two places in southern Guatemala (Tong et al. 2012). This virus could not be propagated, so far, in cell cultures and embryonated chicken eggs which may be due to distinct growth requirements as compared with known influenza viruses. The bat virus, despite its deviation from known influenza A viruses, is potentially capable of exchanging genomic segments through reassortment with human influenza viruses in human cells, and thus, can further contribute to the development of new pandemic or panzootic pool of influenza A viruses. Bats have been considered important animal reservoirs for the emergence of novel viruses not only due to vast species diversity but also because of certain exclusive ecological, biological, immunological and genetic characteristics. Therefore, the isolation of a novel subtype of influenza virus from bats and the known status of bats as reservoir of many animal viruses has raised concerns about the evolution of influenza viruses (Chan et al. 2013).
The sequence data and phylogenetic analysis has revealed relationship and similarities between the HA of the H17N10 bat influenza viruses and the other previously known 16 HA subtypes of influenza A virus. The mean pairwise amino acid sequence identity among the 16 HA subtypes is 49 % (Tong et al. 2012). On an average, amino acid sequence identity of H17 with group 1 HAs and with group 2 was found to be 50 and 38 %, respectively. However, the structure and functions of the presumed receptor binding site of H17 HA was found to be distinct from other 16 HAs. Differences were also observed in the acidic-pH induced membrane fusogenic mechanism in H17N10 viruses (Sun et al. 2013; Zhu et al. 2013). The neuraminidase (NA) gene of this bat virus is substantially distinct from all known influenza NAs. It was further estimated that the known influenza A internal gene lineages diverged after the deviation and divergence of the internal genes from the bat virus had occurred. The atomic structures for N10 NA proteins from three different isolates of bat influenza viruses (H17N10) resolved at ~2.0 Å reflected similarities in the overall N10 structures to each other and to other known influenza NA structures (Zhu et al. 2012). However, considerable differences in the region matching the highly conserved active site of influenza A N1-N9 NA subtypes and influenza B NA were found. The amino acid residues required for NA activity are largely substituted, and the displacement of the two loops (150-loop and 430-loop) has resulted in increased width of the putative active site (Zhu et al. 2012).
It was observed that the atomic structure of N10 largely has resemblance with the structure exhibited by the nine recognised neuraminidases. The N10 lacks canonical NA activity. The residues present in the conserved active site show dramatic alterations resulting in improper binding and cleavage of sialic acid receptors connected at the termini. The intermolecular polar interactions between adjacent N10 molecules of the N10 tetramer occurred due to the participation of an unusual 150-loop (residues147–152) (Li et al. 2012).
The properties and role of N10 have been found to be different from the other nine types of NA proteins of influenza viruses as the neuraminidase activity displayed by the recombinant N10 protein is extremely low or is not present at all. Based on the structural studies and functional analysis, it has been proposed that the N10 protein be designated as NA-like protein for the time being and further investigations are required to reveal its full function(s) (Zhu et al. 2012). An endonuclease activity has been demonstrated in the N-terminal domain of PA of H17N10 bat influenza virus (Tefsen et al. 2013). On the basis of genetic and phylogenetic analysis, an influenza virus isolated from a flat-faced fruit bat (Artibeus planirostris) from Peru has tentatively been classified as H18N11 with a proposal to designate their HA and NA genes as new subtypes H18 and N11 (Tong et al. 2013).
4.9 Evolution of H7N9 Influenza A Virus
Humans in many different countries of North America, South America, and Europe got infected with novel H7 influenza viruses (viz. H7N2, H7N3, and H7N7) between 1996 through 2012 and showed conjunctivitis and mild upper respiratory symptoms. Simultaneous influenza outbreaks in poultry were also reported along with the human infections. During this period of 17 years (1996–2012) none of the humans in China were found to be infected with H7 influenza viruses (Fouchier et al. 2004; Koopmans et al. 2004; Tweed et al. 2004; Nguyen-Van-Tam et al. 2006; Belser et al. 2009; Malik Peiris 2009; Kalthoff et al. 2010; Kuiken et al. 2011). However, beginning from March 2013, a large number of humans in China were found to be infected with a novel strain, H7N9, many of whom died as a result of disease caused by H7N9 strains (Parry 2013; Tang and Chen 2013; Uyeki and Cox 2013; Wen and Klenk 2013; Wiwanitkit 2013; Yang et al. 2013; Wu et al. 2014). The indigenous people of Alaska and Australia have been found to be comparatively much more vulnerable to H7N9 infection (Quiñones-Parra et al. 2014).
The results of many different studies have shown that simultaneous and sequential multiple reassortments involving at least four different sources were responsible for the genesis of the novel avian influenza A H7N9 virus (Fig. 4.5). Domestic ducks and chickens played distinct roles in the genesis of the current H7N9 virus that is presently infecting humans. AIVs of duck origin was the source for the HA gene, and the NA gene came from migratory birds infected with AIVs along the East Asian flyway. Duck H4N9 and H11N9 viruses are considered to be ancestors; however, it is not certain whether these occurred directly or stepwise. Chickens infected with two diverse clusters of H9N2 AIVs contributed the remaining six internal genes. This avian-origin virulent human H7N9 virus emerged after replicating for some time in ducks and chickens that acted as the intermediate hosts. The resulting H7N9 virus began causing outbreaks among chickens in live poultry markets, from which many humans got infected (Bao et al. 2013). The evolution of H7N9 viruses into at least two different lineages is suspected based on the observations of genotypic and potential phenotypic diversities among isolates (Lam et al. 2013; Liu et al. 2013b; Ranst and Lemey 2013; Wu et al. 2013).
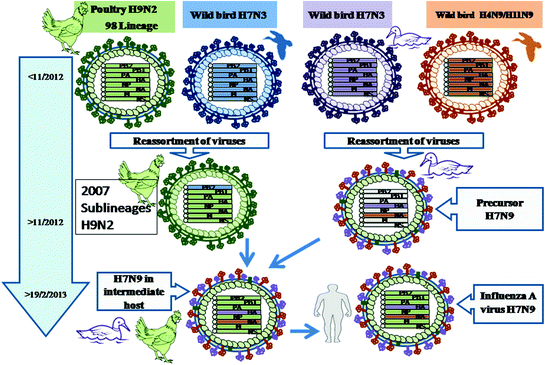
Fig. 4.5
Evolution of influenza A virus H7N9. Reassortment between wild ducks H7N3 and wild bird H4N9, H11N9 in ducks between 2011 and 12 led to the generation of a reassortant designated as Precursor H7N9. Simultaneously, the reassortment between a poultry H9N2 98 lineage and wild bird H7N3 produced at least two lineages of H9N2 in chickens. The reassortant chicken H9N2 reassorted with Precursor H7N9 in ducks and chickens to generate H7N9. After adaptation and replication in the intermediate host (ducks and chickens) this avian-origin influenza A virus H7N9 evolved and was able to infect humans in February 2013
Heterogeneity in the receptor binding properties of the HA of two isolates of avian-origin human-infecting influenza H7N9 isolates was observed. The binding ability of one of the isolate having leucine at 226 residue of HA to the α-2,3-linked sialic acid (avian receptor analogue) was significantly higher, whereas the other isolate, that had glutamine at 226 residue of HA instead of leucine, was able to bind to both α-2,3-linked sialic acid and α-2,6-linked sialic acid (avian and human receptor analogues, respectively) (Liu et al. 2013a; Ramos et al. 2013; Shi et al. 2013a; Shi et al. 2013b). Sequence variations at residues other than 226 on the H7 HA have been reported (Han et al. 2013). There are different opinions whether the evolution of the currently circulating H7N9 isolates would result in a dangerous pandemic strain in future or not. The preservation of predilection by the human H7 virus for avian receptor may limit its further evolution of its conversion into a potential pandemic virus. Although it will be able to infect humans as it has attained some of the receptor binding features observed in pandemic influenza virus (Xiong et al. 2013). The attachment of influenza virus to upper respiratory tract is responsible for the efficient transmission among human. The severity of disease is found to increase if the influenza virus is able to attach to the lower respiratory tract and cause pneumonia. The potential of the emerging H7N9 virus to cause severe disease or of even becoming a future pandemic strain is speculated, as genetic mutants generated in the laboratory were observed to attach to both upper and lower respiratory tracts (van Riel et al. 2013).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


