CHAPTER 59 Equine Protozoal Myeloencephalitis
In 1970, Rooney et al.1 reported 52 cases of focal myelitis-encephalitis in horses from Kentucky and Pennsylvania, with the highest incidence in young Standardbreds. Horses were presented for evaluation of progressive spinal ataxia of one or more limbs. At necropsy, focal lesions of vascular damage, hemorrhage, mononuclear cuffing, gliosis, and neuronal and axonal degeneration were observed in one or more segments of the spinal cord. This report is now thought to be the first published description of equine protozoal myeloencephalitis (EPM).
In 1974, separate reports by Beech and Dodd,2,3 Dubey et al.,4 and Cusick et al.5 described 14 horses in the United States with focal malacia and hemorrhage in the white and gray matter of the brain and spinal cord. Protozoal organisms observed in the central nervous system (CNS) tissues of each horse resembled Toxoplasma gondii but differed in several respects, including an absence of T. gondii antibody responses in affected horses. A subsequent serosurvey of horses revealed that 20% of 1294 serum samples tested by microtitration and indirect hemagglutinin test were positive for antibodies to T. gondii.6 Attempts to induce disease in healthy ponies by oral administration of infective T. gondii oocysts with concomitant corticosteroid injections were unsuccessful,7,8 and the etiologic agent of EPM remained a mystery for almost 20 years.
In 1991 a protozoan apicomplexan parasite was successfully cultured from the spinal cord of a horse with EPM,9 and Dubey et al.10 proposed the name Sarcocystis neurona for the parasite associated with encephalomyelitis in horses in North America. Subsequently, other investigators were able to isolate S. neurona from affected horses,11 and the phylogenetic relationship of the organism to members of the family Sarcocystidae was confirmed based on small, ribosomal ribonucleic acid (rRNA) gene sequence.12 Because of greater than 99.5% homology within a 742–base pair (bp) segment of the 18S rRNA gene, investigators mistakenly suggested that S. neurona was synonymous with Sarcocystis falcatula.13 The opossum is the definitive host for S. falcatula, and birds are intermediate hosts. However, S. neurona is distinguishable from S. falcatula on the basis of its genetic composition, structure, biology, and ability to infect horses, and the two parasites are now recognized as separate species.14–19
ETIOLOGY
Sarcocystis neurona is the most common etiologic agent identified as a cause of EPM, but similar parasites, including Neospora hughesi, have been incriminated as etiologic agents in a few horses.20–25 In this chapter, EPM refers to disease caused by infection of the equine CNS with S. neurona unless specified otherwise.
Sarcocystis neurona has a complex life cycle involving both sexual and asexual stages (Fig. 59-1). The definitive hosts for S. neurona, the opossums (Didelphis virginiana and D. albiventris),26 become infected through ingestion of mature sarcocysts containing bradyzoites in tissues of intermediate hosts, such as the skunk,27 raccoon,28 nine-banded armadillo,29,30 domestic cat,31 and sea otter.32 Bradyzoites are released from the sarcocysts in the gut lumen of opossums and transform directly in the small intestine into male and female gamonts without any replication.33,34 The male gamonts divide into several gametes. An oocyst is produced after the fertilization of the female gamont by the male gamete; the entire process can be completed in the small intestine of the opossum within 24 hours of ingestion of sarcocysts. Oocysts sporulate in the lamina propria, producing two sporocysts, each containing four sporozoites. Fully sporulated oocysts or sporocysts are excreted in the feces of the opossum. Sarcocystis neurona sporocysts from opossum feces are approximately 10 × 8 μm in size.35
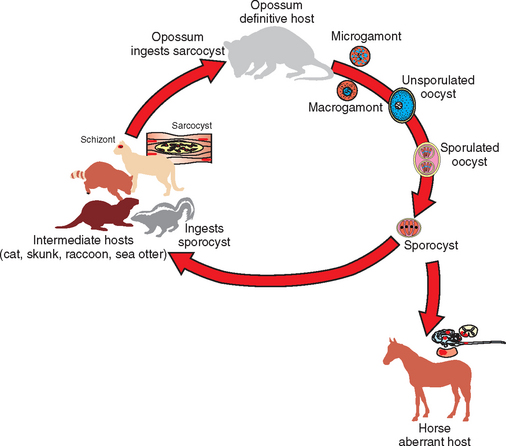
Fig. 59-1 Proposed life cycle of Sacrocystis neurona.
(From Dubey JP: J Vet Parasitol 15:91-102, 2001).
After ingestion of sporocysts by an intermediate host, sporozoites are released, invade the intestinal epithelium, and undergo asexual multiplication in many tissues. The number of asexual generations (schizogony) has not been determined. Studies in interferon-gamma (IFN-γ) knockout mice suggest that S. neurona multiplies in visceral tissues, lungs, and heart before invading the CNS. Sarcocystis neurona multiplies in the CNS and visceral tissues by a specialized form of schizogony, called endopolygeny (Fig. 59-2). In endopolygeny the nucleus becomes lobulated.34 The lobes are connected by chromatin strands and may be arranged in groups. In early stages the uninucleate schizont sometimes resembles a macrophage or a degenerated host cell (Fig. 59-2, C). Finding multiple nucleoli in a nucleus helps to distinguish S. neurona from degenerating host cells. Several merozoites may develop within the same host cell10,34 to become mature schizonts and produce merozoites without leaving the host cell; thus the developmental cycle may be asynchronous. Merozoites are formed centrally or peripherally in the schizont, often around a residual body (Fig. 59-3). Schizonts and merozoites are periodic acid–Schiff (PAS) reaction negative. Mature schizonts in the CNS are up to 30 μm long and may be oval, round, elongated, or irregular in shape.34 The mechanism by which S. neurona is transported is unknown, but it has been proposed that it may reach CNS protected in mononuclear cells (Fig. 59-4).36
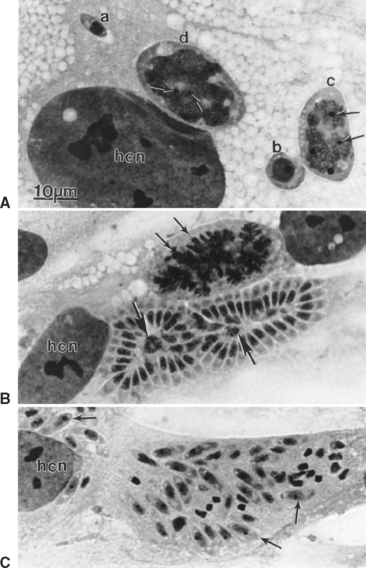
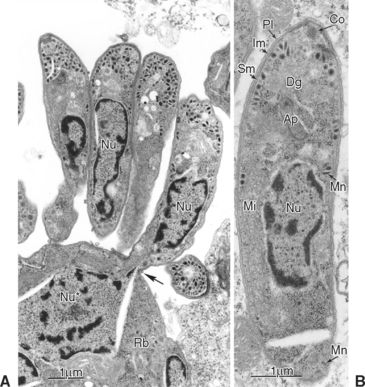
Fig. 59-3 Transmission electron micrographs of Sarcocystis neurona in brain of horse. S. neurona (isolate SN7) was obtained from this horse.31 A, Portion of schizont showing several budding merozoites, one of which still attached (arrow) to the residual body (Rb); Nu, nucleus of merozoite; Nu*, nucleus of schizont still in residual body. B, Merozoite; Ap, putative apicoplast (Golgi adjunct); Co, conoid; Dg, dense granule; Im, inner membrane complex; Mi, mitochondrion; Mn, microneme; Pl, plasmalemma; Sm, subpellicular microtubule.
(From Dubey JP: J Vet Parasitol 15:91-102, 2001.)
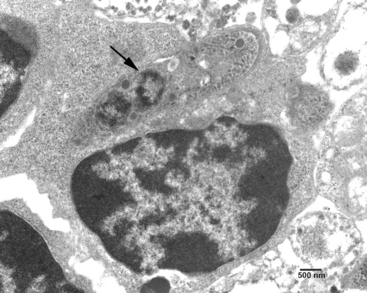
Both neural and inflammatory cells in the CNS may be parasitized. Several hundred merozoites may be present in a single neuron.4 In histologic sections of CNS, individual merozoites are about 3 to 5 μm long and contain a single, centrally located, vesicular nucleus.34
Ultrastructurally, schizonts and merozoites are located in the host cell cytoplasm without a parasitophorous vacuole at any stage of development (see Figs. 59-2 and 59-3). Fully formed merozoites have the same organelles as found in most coccidians (except that rhoptries are absent in Sarcocystis merozoites).37 Merozoites released from schizonts eventually give rise to encysted stages (sarcocysts).31 The earliest sarcocysts consist of a tissue cyst wall enclosing a few round forms, called metrocytes. Metrocytes divide into two organisms that eventually become bradyzoites. Unlike schizonts, sarcocysts are located in a parasitophorous vacuole inside host cytoplasm. It takes approximately 2 months or more for sarcocysts to mature in the intermediate hosts.34 Sarcocysts observed in experimentally infected cats and raccoons were approximately 700 μm in length with a cyst wall 1 to 3 μm thick.31,34,38 Bradyzoites are slender and approximately 5 μm in length. Intermediate hosts do not usually exhibit recognizable clinical signs as a result of S. neurona infection, but encephalomyelitis has been documented in the cat, raccoon, mink, skunk, sea otter, Pacific harbor seal, Canadian lynx, fischer, and dog.39 Horses are considered aberrant intermediate hosts for S. neurona in which only schizonts have been identified with certainty. Raccoons, armadillos, sea otters, skunks, cats, and possibly other mammals are intermediate hosts of S. neurona. 27–31,39–41 In the intermediate hosts, sarcocysts are microscopic; bradyzoites are slender and tiny. The villar protrusions on the sarcocyst walls are up to 2.5 μm long and have microtubules that extend into the ground substance.34
There have been numerous reports of myeloencephalitis in horses associated with Neospora spp. infection.20–23,25,42 A new species, Neospora hughesi, was isolated from infected horses and is considered distinct from Neospora caninum based mainly on genomic analysis of the first internal transcribed spacer (ITS-1) region of the rRNA gene23 and amino acid differences in two immunodominant surface antigens.24
EPIDEMIOLOGY
In North America the opossum (Didelphis virginiana) is the only known definitive host for S. neurona.26 Opossums in North America may be simultaneously infected with several Sarcocystis spp., including S. neurona, S. falcatula, S. speeri, and others.34,43,44 Risk factors associated with the presence of S. neurona sporocysts in opossums include season and higher body condition score.45 Approximately twice as many opossums trapped in spring were positive for sporocysts as opossums trapped in fall or winter. These investigators found no association between sporocyst presence in trapped opossums and age, gender, or the presence of young in the pouch of females.45 A similar study of opossums in southern Michigan revealed that 31 of 206 opossums examined (15%) were infected with S. neurona.46 This study confirmed that summer season was a risk factor for infection, but body condition score was not a factor. A higher frequency of S. neurona infection was observed in adult animals (12.6%) and females (9.2%) than in juveniles (2.4%) and males (5.8%). Multivariate analysis suggested that concomitant infection with other Sarcocystis spp. and presence of young in the pouch of females were risk factors for S. neurona infection in opossums.46
Armadillos,29 raccoons,28 skunks,27 sea otters,43 cats,31,47 and possibly horses48 have been identified as natural intermediate hosts for S. neurona. Sarcocysts isolated from the tongue of wild armadillos were identified as S. neurona, and all 19 armadillos were seropositive for S. neurona by immunoblot analysis.30 Dubey et al.28 demonstrated the presence of S. neurona sarcocysts in muscle of naturally infected raccoons. When these muscles were fed to opossums, S. neurona sporocysts were shed in the feces. Feeding sporocysts to IFN-γ knockout mice resulted in classic protozoal myeloencephalitis. After experimental infection by feeding of S. neurona sporocysts, raccoons developed schizonts and merozoites in many tissues, including the brain, and sarcocysts in skeletal muscle, especially the tongue.38
The raccoon is probably the most important intermediate host for S. neurona in the United States because it is found throughout the country and is often infected with S. neurona. The role of the domestic cat and other intermediate hosts in the natural epidemiology of EPM is controversial, but most researchers consider it unlikely that the cat plays a major role. No S. neurona sarcocysts were observed in muscle sections from 50 free-roaming domestic cats, but S. neurona antibody was detected in 5 of 100 cats.49 Only 5% of 196 feline serum samples submitted to the Michigan State University Animal Health Diagnostic Laboratory for Toxoplasma gondii testing were positive for antibodies to S. neurona. There was no correlation between S. neurona immunoblot results and T. gondii test results, age, gender, or the reason for T. gondii testing.50 Fourteen of 35 cats (40%) from horse farms in southwestern Ohio with prior S. neurona infections in horses on the premises were seropositive for S. neurona. Horses from the same premises had a 93% seropositive rate. In contrast, only 27 of 275 cats (10%) from a spay/neuter clinic in Ohio were seropositive.51
The geographic range of clinical EPM in horses is defined by the range of the opossum, the definitive host of S. neurona. In areas where the opossum is common, approximately 50% of horses are seropositive, indicating exposure to S. neurona.52–58 In central Wyoming and Montana, outside the natural range of opossums, only 6.5% and 0%, respectively, of wild horses are seropositive.59–60 The South American opossum, Didelphis albiventris, is also a definitive host for S. neurona. Approximately 35% of horses in Brazil and Argentina are seropositive to S. neurona by immunoblot analysis.61,62 S. neurona isolates from North America are genetically similar, regardless of the host, but are different from isolates from South America.63
The horse is the most common species in which myeloencephalitis caused by S. neurona infection is identified. In 1998 the U.S. Department of Agriculture (USDA) estimated the incidence of EPM to range from 0.06% in the southern states to 0.43% in the central United States, with a national average of 0.014%.64 Clinical cases of EPM have been diagnosed in native horses of Panama65 and Brazil,66 but the incidence of EPM in seropositive horses in those countries is unknown. Horses in Europe, South Africa, and Asia diagnosed with EPM have invariably been imported from the Western Hemisphere.67–71 Although serologic evidence indicates exposure of other equid species to S. neurona, the only reports of clinical disease in these species are in a single pony and a Grant’s zebra.72–74
Most clinicians consider clinical EPM as a sporadic disease.75 However, prior diagnosis on a premises is a risk factor for diagnosis of future cases of EPM,76 and clusters of disease over a short period have been reported on single farms.65,77 In 1990, Fayer et al.78 reported on 364 histologically confirmed cases of EPM from many areas of the United States. Thoroughbreds, Standardbreds, and Quarter Horses were the breeds most often affected. There were no obvious patterns of infection based on geographic location, gender, or season. Also in 1990, Boy et al.79 reported that the risk for EPM among 82 horses in Pennsylvania was higher in male and Standardbred horses. Neither study had control populations to confirm the significance of the demographic observations.
In 2000, Saville et al.76 reported the results of a case control study of 251 horses diagnosed with EPM at Ohio State University on the basis of positive immunoblot or polymerase chain reaction (PCR) of cerebrospinal fluid (CSF). These horses were compared to a control group of 225 horses with neurologic diseases other than EPM and a control group of 251 horses admitted to the hospital for evaluation of nonneurologic disorders. In the final multivariable logistic regression analysis of EPM horses versus neurologic control horses, factors associated with increased risk of EPM included admission in the fall season, hay storage not secure from wildlife, wooded terrain around the premises, previous diagnosis of EPM on the premises, and a recent (<90 days) adverse health event for the horse.76 Adverse health events included lameness, parturition, accident or injury, management changes, lacerations, surgical procedures, colic, and other medical problems. In the final multivariable logistic regression analysis of EPM horses versus nonneurologic control horses, factors associated with increased risk of EPM included admission to the hospital during the spring/summer/fall season, hay storage not secure from wildlife, use of the horse for racing or showing, age 1 to 5 years, previous diagnosis of EPM on the premises, and a recent adverse health event for the horse. Protective risk factors included age less than 1 year, use for breeding, and observation of birds, but not opossums, on the premises.76
In 2003, Rossano et al.80 reported on a study of EPM in 1121 equids in 98 horse herds in Michigan. Serum samples were tested by immunoblot for presence of antibodies to S. neurona. Samples were stratified based on relative opossum abundance and herd size. Management factors such as age, fly control, wildlife control, and type of feed containment were examined. No single management factor was associated with herd seroprevalence. The authors speculate that it is difficult to restrict exposure of horses to S. neurona on farms located in areas with an abundance of opossums.80
Sarcocystis neurona has been associated with CNS infection and disease in many nonequid mammalian species, including southern sea otters,81–83 Pacific harbor seals,84,85 skunks,72,86 mink,72,87 raccoons,88–90 dogs,39,41 and cats.72,91,92 An unconfirmed S. neurona–like infection has also been reported in a rhesus monkey93 and Canadian lynx.94 The presence of antibodies to S. neurona is a risk factor for myocarditis in southern sea otters.95 Strains of S. neurona isolated from opossums and sea otters are morphologically and molecularly indistinguishable from equine isolates.* The role of S. neurona infection in these species in the natural history of EPM in horses is uncertain at this time.
PATHOGENESIS
The only known method of transmission of S. neurona to horses is through ingestion of sporocysts in opossum feces. Merozoites and schizonts may be observed in a variety of cell types in the CNS of horses with EPM, including neurons, mononuclear cells, and glial cells.4 Only asexual stages of the parasite have been isolated from horses and, in the absence of evidence that sarcocysts form in the muscle of horses, equids are considered aberrant or “dead-end” hosts.
Several investigators have reported clinical signs of disease in one or more horses after experimental infection with S. neurona sporocysts or merozoites.98–102 To date, however, none has successfully fulfilled all of Koch’s postulates by isolating the organism from the experimentally infected horse and subsequently reproducing disease in another horse with that isolate.
In 1997, Fenger et al.98 reported the results of feeding S. neurona sporocysts from opossums to five seronegative foals between 3 and 6 months of age. All foals developed an antibody response to S. neurona in both serum and CSF; four foals exhibited clinical signs of weakness, proprioceptive deficits, spasticity, hypermetria, or ataxia, first observed at 28 to 42 days after infection. Microscopic lesions consistent with EPM were found in three foals, but no protozoal organisms were observed. One foal in which no EPM-like lesions were observed had unilateral, noninflammatory lesions in the cervical and thoracic spinal cord that were consistent with cord compression. Attempts to culture S. neurona from spinal cord samples were unsuccessful.98
In 2001, Cutler et al.102 infected eight seronegative yearling horses orally with sporocysts; concurrently, four of these horses were treated with dexamethasone. All horses were seropositive for S. neurona within 32 days of challenge and were CSF antibody positive within 61 days. Signs of mild to equivocal neurologic disease were observed after 40 days. Mild to moderate multifocal gliosis and neurophagia were observed histologically in the spinal cords of seven horses. No protozoal organisms were identified in any section of the CNS of any of the infected horses. Immunosuppression with dexamethasone did not increase the severity of clinical signs or histologic changes observed in the CNS. Attempts to culture S. neurona from blood, CSF, and CNS tissues were unsuccessful.102
In 2001, Saville et al.101 used a model of transport stress immediately before inoculation with S. neurona sporocysts. Twelve seronegative foals between 4 and 5 months of age were transported for 55 hours. Immediately after transport, three horses were inoculated with sporocysts. Three horses were infected after 2 weeks of acclimation; three horses were acclimatized for 2 weeks and treated with dexamethasone before infection; and three horses remained as untreated negative control horses. Horses infected with sporocysts immediately after transport seroconverted earlier than other horses (9-15 days after infection). All infected horses developed clinical signs of disease as early as 9 days after infection, with the most severe signs in horses infected immediately after transport. One negative control horse developed neurologic signs at 30 days after infection, seroconverted, and had minimal inflammatory CNS lesions at necropsy. One other control horse seroconverted. Histologic lesions observed in infected horses were minimal. All tissue sections from infected horses were negative for S. neurona by immunohistochemistry (IHC), tissue culture, and bioassay in IFN-γ knockout (GKO) mice.101
The experiment was repeated with 24 seronegative weanling horses.103 Six groups of four horses each received varying numbers of sporocysts or placebo. Mild to moderate clinical neurologic signs were observed in challenged horses from all groups, with the most consistent signs observed in the group receiving the highest number of sporocysts. Histologic lesions suggestive of S. neurona infection were observed in 4 of 20 horses fed sporocysts, but parasites were not detected by light microscopy, IHC, or bioassay in GKO mice. Attempts to induce more severe clinical signs by transporting horses a second time 4 to 18 days after infection were unsuccessful.100 All horses seroconverted, and clinical signs were observed as early as 5 days after infection. Parasites were not detected in CNS sections by light microscopy or IHC.100
Sarcocystis neurona has been isolated from the blood and tissues of immunodeficient and immunocompetent horses after experimental infection.99,104,105 The parasite was isolated from the blood of an Arabian foal with severe combined immunodeficiency (SCID) at 21 days after oral infection with S. neurona sporocysts.105 Parasitemia after infection was confirmed in a larger study in which six immunocompetent foals and three SCID Arabian foals were infected either orally with S. neurona sporocysts or intravenously with S. neurona merozoites.99 Foals were stressed by weaning immediately before infection. Despite prolonged parasitemia and persistent infection of visceral tissues (skeletal muscle, cardiac muscle, lung, liver, and spleen), as demonstrated by PCR and culture, SCID horses did not develop neurologic signs after oral or intravenous infection.99 In contrast, although parasitemia was undetectable, four of six orally infected immunocompetent foals developed neurologic signs consisting of ataxia and proprioceptive deficits. Parasites were detectable by PCR or culture in the CNS tissues of immunocompetent foals with neurologic signs. These studies demonstrate that specific immune responses (B and T cell mediated) are required to control parasitemia and infection of visceral tissues of horses, but these responses are often unable to control neuroinvasion and prevent development of clinical signs.99 In 2005, Rossano et al.104 confirmed the presence of parasitemia in immunocompetent horses after oral infection with S. neurona sporocysts by culturing the organism from blood of experimentally infected weanling horses.
Murine models of S. neurona infection provide additional clues to the pathogenesis and immune control of S. neurona in mammalian hosts. Infection of immunocompetent mice is not associated with disease; however, infection of GKO mice, incapable of producing IFN-γ, with either sporocysts or merozoites consistently results in fulminant neurologic disease and death, regardless of the genetic background of the mice.18,106–108 Examination of the CNS of infected GKO mice that die after S. neurona infection reveals inflammatory infiltrates consisting mostly of neutrophils and macrophages, fewer eosinophils, rare multinucleated giant cells, scattered subacute perivascular cuffing, and intralesional protozoa.109 Lesions and organisms are most common in the caudal brain, especially the cerebellum, and equally distributed in white and gray matter of the brain and spinal cord.109
In 2001, Dubey110 described the pathogenesis of S. neurona infection in GKO mice fed sporocysts. Parasitemia was demonstrable by bioassay at 1 to 8 days after infection. Sporozoites were observed in histologic sections of all regions of the small intestine and in cells in Peyer’s patches of a mouse killed 6 hours after ingesting sporocysts. At 1 day after infection, organisms were present in all regions of the small intestine and in mesenteric lymph nodes. Parasites were visible in extraintestinal tissues by 3 days after infection and in the brain within 2 weeks after infection.110,111 Parasites could be found in the brain, liver, lung, heart, and eyes of mice examined at 20 to 62 days after infection.111 Witonsky et al.112 described splenomegaly, lymphadenopathy, mixed inflammatory infiltrate in the liver, perivascular infiltrate in the liver and lung, and interstitial pneumonia in GKO mice at 14 days after infection.
After ingestion of sporocysts from opossum feces, S. neurona replicates to a limited extent in equine gastrointestinal epithelial cells.113 Cell-associated parasitemia provides the parasites with access to visceral and CNS tissues, where subsequent rounds of asexual reproduction occur. After hematogenous spread to the CNS, S. neurona may localize in any area, from cerebrum to spinal cord, but is not found in peripheral nerves. Specific immune responses limit parasitemia and visceral organ infection, but do not always prevent CNS invasion and disease. The exact location of parasite replication in the CNS of an infected horse determines the type and severity of clinical signs that are observed. Some types of immune suppression and stress may increase the likelihood of neurologic disease after infection with S. neurona.
Most information regarding host immune response to S. neurona infection has been gained from murine models of infection. The consistent induction of disease in GKO mice clearly demonstrates the importance of IFN-γ in disease control.18,106–108 IFN-γ is released in the mammalian brain in response to a variety of infectious agents and is considered an essential component of host immune response to CNS infection.114 In C57Bl/6 mice, protection from neurologic disease is mediated by CD8+ T lymphocytes.112,115,116 Mice lacking these cells develop clinical signs of meningoencephalomyelitis with typical histopathologic lesions.115 The relevance of this observation to equine disease is uncertain because of reports that SCID foals (lacking B and T cells) do not develop neurologic disease after infection with S. neurona sporocysts or merozoites, despite persistent parasitemia and widespread replication of parasites in visceral tissues.99
Genetic background also influences development of clinical disease in mice.106,117 Marsh et al.117 reported that C57Bl/6 nude mice (lacking T cells) developed neurologic disease after experimental infection with S. neurona, but ICR SCID mice (lacking B and T cells) did not. This apparently contradictory observation remained unexplained until Ahlgrim et al.106 demonstrated that nude and SCID mice with a C57Bl/6 genetic background develop severe neurologic disease and die after experimental infection with S. neurona, whereas nude and SCID mice on a BALB/c background consistently survive infection without clinical signs. Resistance to clinical disease in BALB/c mice appears to be related to a component of innate immunity other than natural killer (NK) cell function.106,118 When BALB/c SCID mice were infected with S. neurona they failed to develop neurologic disease even when NK cells were depleted. However, these mice did develop fulminant neurologic disease when IFN-γ function was blocked.118 These data suggest that BALB/c mice may have a cell type, other than B cells, T cells, or NK cells, that secretes more IFN-γ than secreted by similar cells in C57Bl/6 mice. The relevance of this observation to equine disease is uncertain, but it may indicate genetic differences in susceptibility to CNS infection and disease in horses.
Relatively little is known about immune responses to S. neurona infection in horses. Two surface proteins of S. neurona, Sn14 and Sn16, are expressed in vivo in the horse and are strong immunogens.119 However, many horses readily develop clinical disease despite the presence of specific antibody, suggesting that humoral responses are not protective. Specific B-cell and T-cell responses limit and control parasitemia and replication of parasites in visceral tissues of horses, but these responses cannot prevent neuroinvasion and neurovirulence.99 Parasites may be able to induce an immunosuppression toward parasite-derived antigens, including suppression of IFN-γ production, facilitating parasite survival in the horse.120 The limited available data suggest that both innate and adaptive immune responses are important for control of infection of S. neurona in horses. Additional information is needed to determine better the nature of protective immune responses and guide production of better vaccines for prevention of disease.
CLINICAL FINDINGS
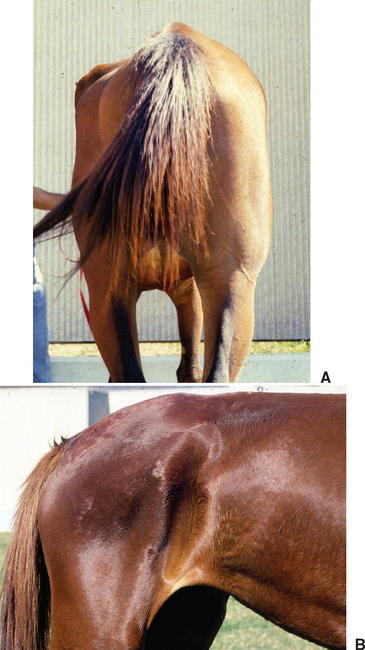
Clinical signs of EPM vary greatly because of the diffuse or multifocal localization of parasites and inflammatory lesions in gray or white matter of the brain, brain stem, or spinal cord.75,121–123 Onset of disease may be acute or insidious, and signs may progress rapidly or remain stable for long periods. Most affected horses are bright and alert with normal vital signs, but focal asymmetric muscle atrophy may be obvious on initial examination (Fig. 59-5). Muscle atrophy is most common in the gluteal or quadriceps muscles but may also mimic sweeney, radial nerve paralysis, or polyneuritis equi.123 Occasionally, affected horses exhibit more generalized, mild muscle atrophy. Complete blood count (CBC) and serum biochemical profile are usually unremarkable.
Initial reports of EPM described horses with progressive neurologic disease that might include ataxia and conscious proprioceptive deficits of one or more limbs, cranial nerve deficits, or cerebral signs.1–5,124 In 1978, Mayhew et al.125 reported that 8 of 32 horses (25%) with EPM had signs of brain disease. MacKay126
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


