CHAPTER 13 Equine Herpesviruses
The equine herpesviruses (EHVs) are highly successful pathogens of all members of the Equidae family worldwide.1–4 Of the nine EHVs characterized thus far, five (EHV-1 to EHV-5) infect the domestic horse, and EHV-6 to EHV-9 are associated with infections in wild equids,5 including asses and zebra.6–9 The EHVs are ubiquitous in both domestic and wild equid populations, and it is likely that the enduring success of the EHV as pathogens results from ancient co-evolution with the Equidae family and adaptation of the virus life cycle to ensure efficient spread within the equid population.
The equid herpesviruses, as with other herpesviruses that infect animals and humans, have sophisticated life cycles adapted to exploit the host animal population and ensure virus persistence. Their life cycles involve infection of multiple cell types in different tissues, with different mechanisms for evasion of the host immune response. Latency, in which recovered horses carry virus in a quiescent (asymptomatic) form for extended periods, is central to the success of these viruses. First, latency provides a mechanism not only for the maintenance of virus in the infected herd, but also for the introduction of virus into new herds through the movement of latently infected horses. Second, latently infected horses form a reservoir of transmissible infection through reactivation, resulting in infection of new, susceptible, in-contact horses.
In domestic horse populations the most important and most studied of the EHVs are the alpha herpesviruses EHV-1 and EHV-4. These respiratory pathogens are also responsible for abortion and neurologic disease. EHV-3 is an uncommon venereal pathogen and is not discussed in this chapter (see Chapter 8). The clinical importance of the gamma herpesviruses EHV-2 and EHV-5 is less clearly defined, although some evidence suggests that EHV -2 and EHV-5, in certain situations, may be clinically important as respiratory and ocular pathogens (see Chapter 10). There has been speculation that these viruses may act as immunosuppressive agents, predisposing to other viral infections, or may be involved in persistent, chronic fatigue syndromes.
The EHVs have a major economic and welfare impact on all sectors of the horse industry worldwide through their direct clinical effects on the horse, including respiratory disease, abortion,10,11 and paralysis,12 and through their effects on the horse industry, including interference with horse movement for breeding and competition. The first of the equid herpesviruses to be described were EHV-1 and EHV-4.13–15 These viruses are closely related, but genetically and phenotypically distinct, and were originally designated as subtypes 1 and 2 of EHV-1.16–20 These two viruses have been the subject of a major international research effort over the past five decades. This research has produced a wealth of fundamental virologic and immunologic knowledge about their pathogenesis and the interactions between these viruses and the horse.3 This information in turn has allowed effective management control programs to be devised (embodied in the United Kingdom in the Horserace Betting Levy Board’s Code of Practice; www.hblb.org.uk). From the outset, EHV research has been directed toward vaccine development; the first EHV-1 vaccines were used in 1961,21 and there are now a variety of killed and live commercial EHV vaccines available in Europe and North America.
ETIOLOGY
Relevance of Virology to Clinical Practice
Advances in the understanding of the molecular biology and virology of the equid herpesviruses have greatly improved our understanding of their pathogenesis,1,3 the immune responses required to control virus replication and spread in the host,22 and critically the identification of key immunologic viral targets.23,24 This information is a prerequisite for effective vaccine design.
Virology Overview
EHV-1 and EHV-4 are alpha herpesviruses (members of the Alphaherpesvirinae subfamily) and belong, along with bovine herpesvirus-1, suid herpesvirus-1, canine herpesvirus-1, felid herpesvirus-1, and Marek’s disease virus (MDV), to the Varicellovirus genus, the prototypic virus of which is the human pathogen varicella-zoster virus (VZV).25 The varicelloviruses share some similarities with the herpes simplex viruses HSV-1 and HSV-2, the cause of cold sores and genital herpes, respectively, but are genetically and phenotypically distinct. Indeed, the varicelloviruses are sufficiently different to merit care in extrapolation of data from the simplex viruses to the EHV, as with the use of the antiviral agent acyclovir to treat EHV-1 neurologic disease (see Treatment).
EHV-1 and EHV-4 are closely related26,27 but genetically and antigenically distinct with different disease profiles. EHV-1 is principally a pathogen of the domestic horse, but there is serologic evidence of occasional infection of domestic cattle and captive camelids and cervids. In contrast, EHV-4 is an exclusive pathogen of the domestic horse. In the laboratory, some aspects of EHV-1 pathogenesis can be modeled with limited success in mice28–30 and hamsters,31,32 but this has not proved possible for EHV-4.
Within the Varicellovirus genus there is a spectrum of cellular tropism from principally neurotropic viruses (e.g., VZV) to viruses that are principally lymphotropic (e.g., MDV). Equine herpesviruses types 1 and 4 lie midway along this spectrum, being both neurotropic and lymphotropic during their life cycle (see Pathogenesis). A key feature of the life cycle of EHV-1, distinguishing it from EHV-4, is that it efficiently infects a variety of cell types,33 including respiratory epithelial cells,34 endothelial cells,35 neuronal cells,36 and lymphoid cells.37,38 EHV-4, on the other hand, has a tropism principally restricted to epithelial cells and neuronal cells and has limited potential for infection of endothelial and lymphoid cells.
The gamma herpesviruses EHV-2 and EHV-5 are principally lymphotropic, establishing functional latency in these cells.39,40 Although viral deoxyribonucleic acid (DNA) has been detected in trigeminal ganglia,41 reactivation from this site has not been demonstrated. EHV-2 and EHV-5 have typical Gammaherpesvirinae genome structures, with close similarity to saimiri and Epstein-Barr viruses.42 They have different disease profiles from EHV-1 and EHV-4 and share no cross-protective antigens with these viruses.
Viral Genomes
In the short time since the complete genome sequences of EHV-1 and EHV-4 were determined,43,44 partial or complete virus genomes can now be sequenced comparatively easily, and sequencing entire genomes of EHVs of interest is now a practical prospect. The availability of genome sequence, together with the molecular techniques and reagents to manipulate individual genes or groups of genes, has revolutionized understanding of EHV-1 gene function and regulation. The genomes of several EHV-1 isolates, including high-virulence and low-virulence strains, have been cloned as bacterial artificial chromosomes into Escherichia coli, providing a greatly improved means of studying viral gene function.45–47 Transposon mutagenesis, a well-established technique in microbial genetics, has been successfully applied to EHV-1, providing a rapid means of screening the entire genome for virulence genes.
EHV-1 and EHV-4 possess linear double-stranded genomes and share the same overall genome conformation with a unique long region (UL) joined to a unique short region (US) that is flanked by an identical pair of inverted repeat regions, the terminal repeat (TR) and internal repeat (IR) regions43,44 (Fig. 13-1). Genes located in the repeat regions are thus represented twice in the genome. Both genomes are of similar size and share extensive sequence homology but are genetically (and phenotypically) distinct viruses. The EHV-1 genome is 150 kilobases (kb) in size and encodes 76 open reading frames (ORFs; genes), of which 63 are located in the UL, nine in the US, and four in the repeat (IR) regions (Table 13-1). The EHV-4 genome is slightly smaller (145 kb) with 76 genes, each of which has a direct positional and sequence counterpart in EHV-1. The EHV-2 genome is larger (184 kb) and contains 79 ORFs, of which 77 are predicted to code for proteins.42
Table 13-1 Open Reading Frames (ORFs) of Equine Herpesvirus Type 1 (EHV-1) and Predicted Functions of Their Gene Products
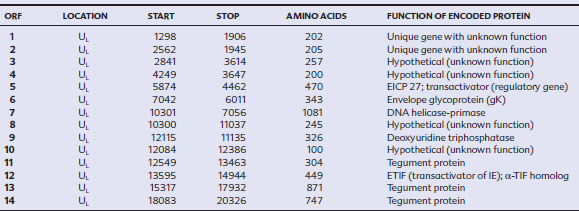
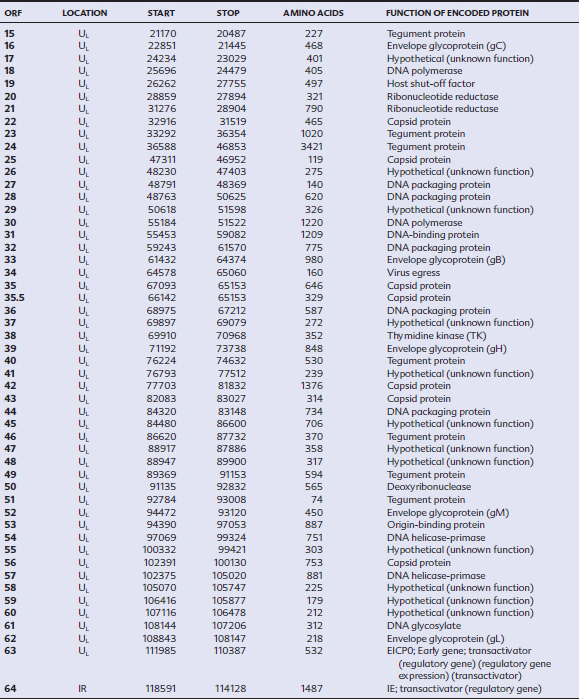
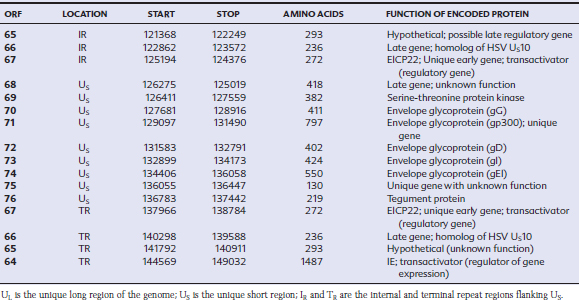
Both EHV-1 and EHV-4 possess five unique genes (numbers 1, 2, 67, 71, and 75) that have no homologs in other sequenced herpesviruses and have been the subject of considerable research in the expectation that their products play pivotal roles in pathogenesis. Although the roles of these unique genes have not been completely elucidated, studies in vitro and in mice have failed to identify distinctive roles in pathogenesis.48,49
Viral Replication
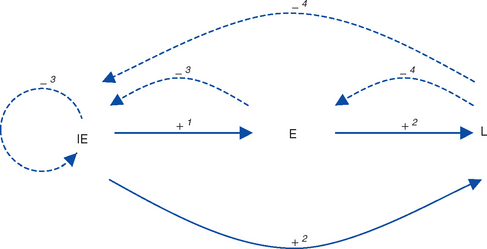
The key feature of the molecular biology of EHV-1 and EHV-4 is that gene expression is tightly ordered into a highly controlled cascade.50 The result is that the horse’s immune system is exposed to virus gene products in a sequential fashion, providing specific opportunities for immunologic intervention. One complete cycle of virus replication (the lytic cycle) takes approximately 20 hours, during which a well-ordered progression of events occurs: attachment to the host cell membrane, membrane fusion and penetration, translocation of viral DNA to the nucleus, viral DNA replication, viral protein synthesis, capsid assembly, egress from the nucleus, envelopment and egress from the cell, and death (lysis) of the cell as progeny virions are released. During these different stages of infection, viral gene transcription and regulation is sequentially regulated into three distinct phases: immediate early (IE), early (E), and late (L) (see Virus Proteins).50–53 Gene products from each phase have regulatory roles that, acting in concert with host cell proteins, upregulate (“switch on”) expression of other phases while downregulating (“switching off”) their own phase (Fig. 13-2).
Lytic and Latent Infection Cycles
On entry to host cells, the virus enters either a lytic cycle or latent cycle of infection. These two pathways appear to be independent, to occur simultaneously, and to diverge immediately after viral entry in the cell. Virus replication is not required for the establishment of latency. The lytic cycle, as previously described, results in release of new virus particles from the infected cell. During the latent cycle of infection, viral DNA translocates to the nucleus, but the transcription and translation cascade of all gene classes is blocked, with the exception of limited transcription in the region antisense to the IE gene. This results in expression of the EHV latency-associated transcript (LAT).54–56
The mechanisms that control entry into lytic and latent infection cycles are not well understood. In the nervous and lymphoid systems, it appears that the two infection cycles occur in parallel, principally within neurons of the trigeminal ganglion and CD8+ T lymphocytes.36,57,58 Although the genetic mechanisms responsible for suppression of gene transcription and translation in latency have not been fully elucidated, it seems unlikely to be entirely caused by antisense repression of IE gene transcription by LAT messenger ribonucleic acid (mRNA).
During latency, the genome exists in multiple copies in a continuous (circular) episomal form in the nucleus of infected cells. Latently infected cells do not express viral antigens and are thus not detectable immunologically. Latently infected cells represent a subset of neurons and CD8+ lymphocytes.59
Periodically, the latent genome undergoes reactivation, during which a “reverse trip” occurs in which unenveloped virus capsids are assembled within latently infected cells before translocation to the respiratory epithelial surface, where they undergo envelopment and become infectious virus particles. Once delivered to the respiratory epithelial surface, virions undergo one of two fates: (1) neutralization by local mucosal immunity or (2) establishment of infection and a repeat of the events that occurred during primary infection, with shedding of infectious virus in respiratory tract secretions and possibly development of viremia60 (Fig. 13-3). The precise details of this reverse trip from neurons of the trigeminal ganglion36 and circulating T lymphocytes59 are not fully understood. It is assumed that the transfer of reactivating virus from lymphocytes to the respiratory epithelium involves expression of viral proteins on the surface of reactivating lymphocytes to allow cell-cell fusion and direct virus transfer. This process may also occur within the uteroplacental unit and the central nervous system (CNS), providing a mechanism for disease recurrence with reactivation.3
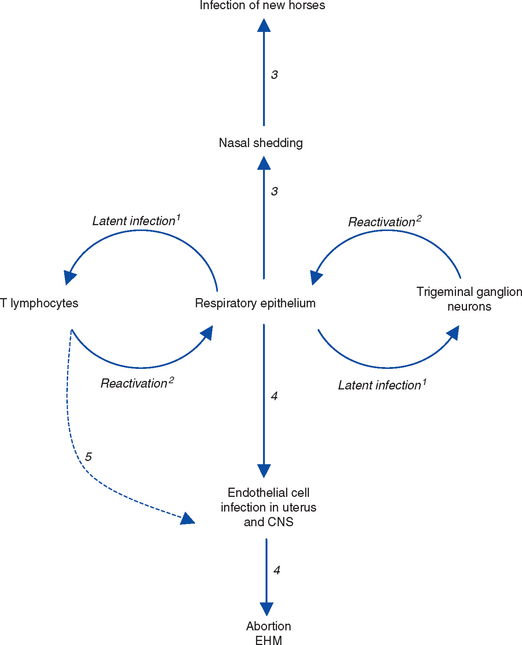
At a cellular level the molecular, cellular, or other events that control the switch from latency infection to reactivation are unknown. Reactivation involves a switch from latent to lytic cycle that is initiated by transcription and translation of the IE gene, inducing the lytic gene cascade as seen during primary infection. It is not known whether initiation of transcription requires a specific trigger, possibly trans-activation of the IE promoter by viral, cellular, or exogenous factors, or whether it occurs spontaneously. The IE promoter can be transactivated by a variety of viral and cellular proteins, including proteins encoded by EHV-2,61 although the biologic significance in relation to reactivation is not known.
Whatever the mechanism of reactivation, it is likely that the outcome of most reactivation events, which may occur frequently at the cellular level, is neutralization of reactivating virus by mucosal immune responses, and thus prevention of virus shedding from the respiratory tract and development of viremia. Reactivation only becomes detectable at horse level, by nasal shedding or viremia, when the host immune response is compromised either by treatment with corticosteroids60 or other immunomodulatory compounds or by a variety of management stressors,62 including transport, illness, and hospitalization. It is not known how frequently horse-level reactivation and shedding actually occurs, but it is sufficiently frequent to maintain these viruses in the global horse population.
Viral Proteins
The virus encodes a single IE gene, 55 E genes, and 20 L genes. The 76 gene products have a variety of different functions, giving the EHV surprising complexity and comparatively sophisticated life cycles, features that greatly complicate attempts to understand pathogenesis and devise control measures. Functions have been assigned to the majority of viral proteins. At least 30 are associated with the virion, of which six form the capsid,63 12 are associated with the tegument64,65 (located between the capsid and the envelope), and a further 11 are glycoproteins66 anchored to, and projecting from, the envelope. The remaining proteins are involved with viral replication functions, including DNA replication enzymes; regulation of the viral gene cascade; and virus egress from infected cells. The regulation of the sequential cascade of viral genes into IE, E, and L phases50 is controlled by six genes whose gene products act as transactivators and transcriptional regulators/suppressors: the IE gene (gene 64), four E genes (EICP 22, 27, 0 and TR2), and one L (ETIF) gene. Five of these genes (IE, EICP22, EICP27, TR2, and ETIF) are essential for virus replication in vitro.
The single IE protein is extremely important from an immunologic perspective because it is the first virus expressed by infected cells and, in some horses at least, contains a dominant cytotoxic T lymphocyte (CTL) epitope, making it a key vaccine target.24,50,51,67–74 The 1487 amino acid IE protein is essential for virus replication and is a vital regulatory protein,75 repressing its own promoter and transactivating expression of early and late gene promoters. It possesses different functional domains, including a DNA-binding domain responsible for binding its own promoter, a domain responsible for translocation of the IE protein to the infected cell nucleus, and a domain that binds transcription factor IIB (TFIIB) from the infected cell.76–78
Four E proteins (EICP 22, 27, 0, and TR2) have regulatory activities. EICP22 and 27 (gene 5) function synergistically with the IE protein to transactivate E and L genes, and EICP0 (gene 63) is also a powerful transactivator but is antagonistic to the IE protein and competes for the cellular transcription factor TFIIB.47,52,77,79–85
The late protein regulatory protein ETIF53,86 is the product of gene 12 and is the EHV-1 equivalent of the herpes simplex virus alpha trans-inducing factor (α-TIF), a key transactivator that binds upstream of the IE gene and transactivates the IE promoter. ETIF is required for cell-to-cell spread; ETIF mutants produce small plaques in cell culture, and although capsids are produced, envelopment in the cytoplasm does not occur.
Glycoproteins
The 11 envelope-anchored surface glycoproteins of EHV-1 and EHV-4 play key roles in pathogenesis, including host specificity and cellular tropism.66,87 They mediate viral attachment to and entry into cells as well as fusion of infected cells and direct cell-to-cell spread of virus. They are the principal viral immunogens, at least for humoral immunity, and are the major targets for virus-neutralizing antibody. Five glycoproteins (gB, gD, gH, gK, gL) are absolutely required for virus replication in tissue culture and are termed essential. The remaining six (gC, gE, gG, gI, gM, gp300) are not required for replication in vitro and are termed nonessential, a designation that does not translate to the in vivo situation because deletion mutants in these glycoproteins are viable but less virulent. Although convalescent horse sera recognize all 11 glycoproteins, three (gB, gC, gD) are “immunodominant,” that is, are the principal viral antigens recognized by the equine host. Further features of gB, gC, and gD that have made them prime vaccine candidates are (1) they are protective in mice; (2) gB, gC, and gD deletion mutants are nonpathogenic in mice; (3) gB and gD are involved in cell-cell fusion and spread of virus; and (4) gD is involved with virus entry into cells and appears to be responsible for cellular tropism and host cell specificity.88–99 The functions of the other glycoproteins are less well characterized; gE and gI are involved in cell-cell spread of virus, gK in cell-cell spread and virus egress, and gM in cell penetration and cell-cell spread46,68; gH is partially protective in mice.100
EPIDEMIOLOGY
The principal reservoir of infection for EHV is latently infected horses. Environmental transmission is of most importance during outbreaks when horses are kept in close confinement and probably plays a minor role in maintenance of these viruses in the horse population. Environmental persistence of EHV is short, estimated to be less than 7 days in most conditions, with a maximum survival of 35 days.101 The viruses are labile and easily inactivated by heat and disinfectants. Serologic surveys suggest that most adult horses have been exposed to EHV-1, -2, -4, and 5. Epidemiologic studies of stud farms suggest that infection is acquired within the first few weeks or months of life, generally before or just after weaning, from adult mares that asymptomatically shed virus102–105 (Fig. 13-4).
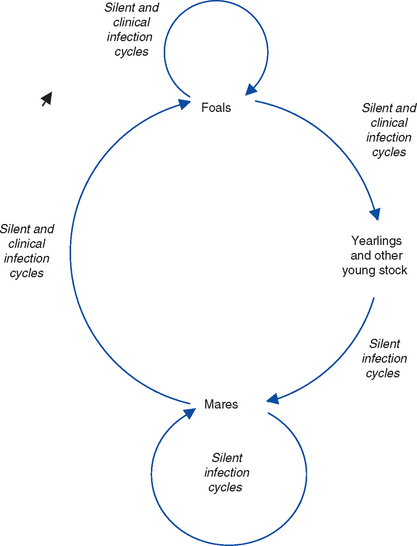
It is not clear whether the maintenance of endemic infection is caused by cycles of subclinical infection within the mare population or reactivation of endogenous latent infection in individual mares. The life cycles of the EHV share four main features: (1) early and widespread infection of young horses, (2) a widespread carrier (latent) state in adult horses, (3) transmission from latently infected adults to new generations of young stock, and (4) widespread “silent” adult-adult, adult-foal, and foal-foal endemic infection cycles of transmission.
Latency
Latency and reactivation are key features of the epidemiology of EHV-1 and EHV-4 infections and are responsible for the ubiquitous distribution of these viruses in the horse population. The large majority of recovered horses carry latent EHV infections for extended periods, possibly for life.57,58 Latency almost certainly also plays a key role in the biology of EHV-2 and EHV-5, since the majority of adult horses and almost all foals harbor latent infection in circulating lymphocytes. EHV-1 and EHV-4 enter into a latent state in the lymphoreticular system, in circulating and lymph node CD8+ T lymphocytes, and in neurons within the trigeminal ganglia.36 EHV-2 and EHV-5 establish functional latency in circulating lymphocytes and trigeminal ganglia, from which virus can be reactivated in vitro.106,107 Viral DNA can also be detected in other locations,108 including the trigeminal ganglia,41 but it is not clear whether virus is capable of undergoing reactivation from these other sites. These latently infected cells form, by means of periodic reactivations that result in shedding of infectious virus from the host, a transmissible reservoir of infection that maintains all the EHVs in the horse population.
Reactivation from Latency
Periodically, latently infected horses experience reactivation episodes, during which infectious virus is shed into respiratory tract secretions, with the potential for infecting other, susceptible horses. Reactivation of latent EHV-1 and EHV-4 infections from horses has been observed in field situations after transport, handling, rehousing, and weaning, and reactivation has been achieved experimentally in horses by treatment with high doses (10 times therapeutic doses) of corticosteroids.36,109 The frequency with which reactivation occurs in response to the stresses imposed by the modern horse industry is unknown. Reactivation is generally asymptomatic, resulting in “silent” shedding of virus, thus providing a mechanism for maintenance of endemic infection cycles and the apparently unexplained appearance of disease in closed populations of horses.
Abortion or neurologic disease may be the result of local reactivation of virus, that is, reactivation of EHV-1 within blood vessels of the uterus, placenta, or CNS, resulting in endothelial cell infection and thrombo-ischemia in those organs.110–113 In this situation, disease occurs without prerequisite respiratory infection, nasal virus shedding, or viremia. Regardless of the sites from which reactivation occurs, it is likely that latency and reactivation are important in the epidemiology of EHV-1 abortion and neurologic disease. The majority of natural EHV-1 abortions occur singly,114 rather than as “abortion storms,”115 implying that abortion may have resulted from reactivation of endogenous latent virus rather than from a newly acquired respiratory infection. Such reactivations of latent EHV-1 may also explain abortions that occur many weeks or months after infection, beyond the period of viremia that follows. It is not clear whether latency is also important in the epidemiology of neurologic disease. However, it is logical to assume that endogenously reactivated virus may be responsible for isolated neurologic cases or for seeding virus into groups of horses where outbreaks of neurologic disease occur.
PATHOGENESIS AND PATHOLOGIC FINDINGS
Respiratory Tract
Following inhalation of virus or contact with infected fomites, both EHV-1 and EHV-4 replicate in upper respiratory tract epithelial cells. After experimental infection, virus can also be transiently recovered at low titer from lower respiratory tract (tracheal and bronchoalveolar) lavage samples. Virus replication causes death of epithelial cells, resulting in epithelial erosions. EHV-1 quickly spreads to cells in the underlying lamina propria, with infected (viral antigen-expressing) endothelial cells, lymphocytes, and monocytes detectable in respiratory tract–associated lymph nodes within 48 hours.116,117 From these sites, virus-infected lymphocytes enter the circulation, resulting in a CD8+ T lymphocyte–associated viremia that disseminates virus widely, including into the uterine and CNS vascular endothelium.118,119 In parallel, EHV-1 also rapidly gains access to neurons of the trigeminal nerve, reaching the trigeminal ganglion by 48 hours after infection, establishing latency in trigeminal ganglionic neurons.36 Experimentally, EHV-1 shedding occurs from the respiratory tract for up to 14 days, and viremia persists for up to 21 days.120 In the field, however, shedding and viremia may be more transient and intermittent, making detection difficult in the later stages of infection.
The detailed pathogenesis of EHV-4 infections has not been elucidated. The initial mucosal phase probably closely parallels that of EHV-1, with infection of the respiratory tract and its associated lymphoid system. EHV-4 DNA persists in the trigeminal ganglion121 and circulating lymphocytes58 after recovery from infection, but infectious virus has not successfully been reactivated from these sites in vitro. Most isolates of EHV-4 have low endotheliotropism, do not establish viremia, and therefore do not cause abortion or neurologic disease.33,122 The duration of nasal shedding of EHV-4 is usually more transient than for EHV-1.
Viremia
Viremia disseminates virus to the uterus123 and CNS and is therefore a prerequisite for these diseases. However, not all viremic pregnant mares abort, and only a very small minority of viremic horses develop neurologic disease. Local reactivation can also be responsible for these diseases without detectable viremia (see Latency and Reactivation). Viremia also delivers virus to other organs, detectable by polymerase chain reaction (PCR) assays, but does not result in clinically apparent organ dysfunction. Viremia is cell associated, primarily within CD5+/CD8+ T lymphocytes,124,125 and free virus is rarely detected in the blood. Lymphocytes do not support lytic infection in vivo; virus can be liberated from these cells using only co-cultivation (in vitro reactivation) assays. Lymphocytes are susceptible to infection in vitro only after mitogen stimulation,126 which also increases the efficiency of reactivation in vitro.59 Viremia seldom occurs with EHV-4 infection, although some “high-virulence” isolates capable of inducing viremia exist, and EHV-4 DNA can be detected in circulating lymphocytes after infection.127
Uterus
The pathogenesis of EHV-1 abortion involves virus translocation from the circulation into the placental unit and induction of uterovascular lesions.128 Infection of endothelial cells in the pregnant uterus causes a vasculitis that affects small arteriolar branches in the glandular layer of the endometrium at the base of the microcotyledons.113 If these endometrial vascular lesions are widespread, the fetus may be aborted before detectable transplacental spread of virus has occurred.129,130 Virologically negative abortions129 in earlier EHV-1 challenges had been assumed to be caused by maternal stress or pyrexia, and the incidence of this type of abortion after spontaneous field infections is not known. However, in a survey of 241 abortions in the United Kingdom between 2001 and 2003,130 nine were “typical” EHV-1 abortions (i.e., placenta and fetus were virus positive), whereas six were “atypical,” and there was no detectable fetal infection by either PCR or immunohistochemical methods.131 The susceptibility of uterine endothelial cells to infection with EHV-1 is low in early pregnancy compared with late pregnancy, and any potential association of EHV-1 infection with early embryonic death and resorption has not been investigated.
The pathogenesis of abortion caused by less virulent isolates of EHV-1 is not as clear,132 because these isolates appear to have a reduced affinity for endothelial cells; however, abortion is also caused by vascular lesions and thrombo-ischemia. The rare cases of abortion that follow infection with EHV-4 are also likely to involve the capacity of certain isolates of EHV-4 for replication in uterine and fetal endothelial cells.
Central Nervous System
The pathogenesis of EHV-1 neurologic disease also involves vasculitis and thrombo-ischemia following endothelial cell infection.133 There is little evidence for lytic virus infection of neurons or direct viral neuropathology,134 and virus has rarely been isolated from the CNS. Affected horses demonstrate sites of vasculitis with virus antigen expression in the brain and spinal cord, with or without local hemorrhage and thrombo-ischemic necrosis.136,137 Neurologic clinical signs therefore result from ischemic death of nervous tissue, and the virus thus causes a myelopathy, encephalopathy, or myeloencephalopathy, depending on which regions of the CNS are affected. EHV-1 neurologic syndromes are therefore generally referred to as equine herpesvirus myeloencephalopathy (EHM).
Virulence
There are wide variations in virulence among different isolates of EHV-1 and EHV-4. High-virulence EHV-1 strains, such as Ab4 and Army-183, are endotheliotropic, efficiently cause viremia, are abortigenic, and after experimental inoculation of horses, cause high rates of abortion and neurologic disease. Other isolates, such as V592, are less virulent, have reduced endotheliotropism, cause only “low-level” viremia, and rarely cause abortion or neurologic disease.33,132 The availability of genome sequences for different EHV-1 strains has allowed identification of sequence variations that correlate with specific phenotypes. Small differences at genome sequence level account for marked phenotypic differences. For example, Ab4 and V592 exhibit only 0.1% difference in sequence (150 bases in 150,000). These are mostly single-base changes, causing coding changes in 31 genes. Gene 68 (US2) shows the highest variation rate (2%) and is now used as a phylogenetic marker for epidemiologic analysis. However, no relationship appears to exist between the phylogenetic groups generated by this method and the pathogenic potential of strains.
Immunology
Natural or experimental infection induces a solid but transient protective immune response to EHV that protects against reinfection for 3 to 6 months. During this time, there is clinical protection and sterile immunity (i.e., exposure to virus does not induce nasopharyngeal virus shedding or viremia). Exposure of mares early in gestation protects them from abortion in the later, susceptible stages of gestation. Furthermore, mares that have aborted in 1 year usually do not abort in subsequent years.181
The life cycle of EHV-1, and to a lesser extent EHV-4, is complex and involves infection of multiple cell types.3 The initial mucosal colonization and replication phase of infection is rapidly followed by an invasion phase, during which virus enters the lamina propria and gains entry into blood vessels and lymphatics through endothelial cell infection, and a distribution phase, where infected lymphocytes disseminate virus throughout the horse. In parallel, latency is established. It is now clear that immune control of pathogenesis requires an integrated, multicomponent immune response, and that there are unlikely to be simple correlates of protective immunity. This is in marked contrast to “hit and run” virus pathogens with comparatively simple pathogenesis, exemplified by equine influenza viruses, for which measurement of the humoral immune response provides a simple and accurate measure of disease susceptibility and resistance.
Infection induces strong systemic humoral immune responses characterized by an initial, rapid, but short-lived (<3 months) immunoglobulin M (IgM) response, followed by a slower-onset but longer-lived (>12 months) immunoglobulin G (IgG) response (see Diagnosis). The principal IgG isotypes produced are IgGa, IgGb, and IgGc. Only small quantities of immunoglobulin A (IgA) are found in the circulation in convalescent sera. In diagnostic laboratories, these responses are measured using the complement fixation (CF) test for the short-lived (IgM) antibody response and the virus neutralization (VN) test for the longer-lived (IgG) antibody response. Systemic humoral immune responses alone are not sufficient to protect horses from infection. VN antibody titers do not correlate with protection from infection. High VN titers are associated with reduced nasal virus excretion but do not influence the duration of viremia or the frequency of viremic cells in the circulation,182 presumably because viremic cells express few, if any, virus antigens and are therefore not recognized as targets by the immune system.183 Infection induces mucosal immune responses characterized by mucosal IgA production. In contrast to VN antibody, mucosal IgA titers do correlate with protection from infection.184 The IgA response is short lived after a single infection but persists longer with subsequent infections. Mucosal IgA is an important first line of defense against EHV-1 infection. It is known to be neutralizing in vitro, although its function in vivo has not been fully characterized.
Infection induces both tissue (mucosal and lymphoid) and circulatory cellular immune responses mediated by CD8+ cytotoxic T lymphocytes (CTLs).185–187 Understanding of the cellular immune response has advanced rapidly in the last 10 years, and there is abundant evidence that CTL responses are not only central to recovery from infection but also provide a correlate of protective immunity.22,188 CTLs are the effector arm of the cellular immune response and kill infected cells that present virus antigens on their surface in the context of major histocompatibility complex (MHC) class 1 (ELA-A) molecules; that is, CTL responses are MHC-1 (ELA-A) restricted. The CTL response is directed by “professional” antigen-presenting cells (dendritic cells and macrophages). These cells are also MHC-1 restricted and work in concert with the Th1 subset of CD4+ T helper cells and an associated panel of cytokines, including interleukin-2 (IL-2), interferon-gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α).24,189 The polymorphic nature of ELA-1 molecules in the horse means that individuals vary in their CTL responses to particular virus antigens. As with other herpesviruses, MHC-1 expression is downregulated on the surface of infected cells, thus providing a means of immune evasion by the virus.
EHV-1 and EHV-4 infections cause changes in leukocyte populations in infected horses characterized by leukopenia followed by leukocytosis. The initial leukopenia consists of lymphopenia and neutropenia, with an initial depletion in CD8+ cells, and occurs between days 7 and 13 after infection. This is followed by a leukocytosis, consisting principally of a lymphocytosis, that continues to day 21 to 28 after infection. EHV-1, but usually not EHV-4, infection induces a cell-associated viremia that is associated mainly with infection of CD8+ cells, although a small proportion of monocytes and possibly CD4+ cells also become infected. In vitro, these cells are refractory to infection unless stimulated by mitogens. Both CD8+ and CD4+ cells release IFN-γ, an indirect marker of cytotoxic activity, in response to EHV-1 infection in vitro, suggesting both cell types are responsive to EHV-1.24,189
CTL responses to EHV were initially measured using a bulk lymphoproliferative assay.186 Although lymphoproliferative responses increased after infection, there was no clear correlation with protection. The assay was refined by measuring the frequency of CTL precursors (CTLp) by limiting dilution analysis (LDA).188 For the first time, this provided a correlate of protective immunity: high numbers of CTLp correlated with protection, whereas horses with low CTLp numbers were susceptible to infection. High frequencies of circulatory CTLp also correlate with protection from abortion in the face of EHV-1 challenge.22
The EHV-1 antigens that act as CTL targets have been mapped, providing valuable information on which virus antigens stimulate cellular immune responses. A variety of viral proteins have been mapped, including IE, several glycoproteins (e.g., gB, gC, gD), structural proteins, and proteins involved in replication. The IE protein, the first virus protein to be expressed in infected cells, is the most efficient CTL epitope. However, the ability of this protein to induce CTL responses varies with MHC haplotype; efficient responses are seen only in horses with the ELA-A3.1 haplotype.23 A universally immunodominant CTL epitope, the functional equivalent of gB, gC, and gD for the humoral immune response, seems unlikely, but a panel of viral proteins constructed around the IE protein may fill this role.
Immune Evasion
The changes that occur in blood and pulmonary leukocyte populations after EHV-1 infection, including the lack of viral antigen expression on the surface of infected leukocytes, have led to the recognition that EHV-1, as with many of the herpesviruses, modulates the horse’s immune response. Because cytotoxicity against EHV-1 proteins is MHC-1 restricted, it is not surprising that EHV-1 produces proteins that interfere with MHC-1 presentation of virus antigens as a means of evading the horse’s immune response to infection.190 Whether this immunomodulation represents immunosuppression
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


