Fig. 11.1.
l-Methionine (MET) decreases spine density on both the apical and the basilar dendrites. Mean spine density ±SE shown here for a distance of 50–100 μm from the pyramidal cell body located in frontal cortex layer III. Mice were treated for 7 days with VEH (saline twice daily s.c.) or MET (5.2 mmol/kg twice daily s.c.). VEH, 13 mice; MET, 12 mice. The difference in mean spine density was assessed by unpaired t-test, ** p < 0.01.
Weaver et al. (72) administered MET for 7 days to offspring of high licking and grooming rat mothers. A microarray analysis of gene expression indicated that only a small fraction of genes analyzed in the hippocampus were affected by repeated administration of MET, and RELN was one of the few genes affected. The upregulation of reelin in the hippocampus of offspring of high licking and grooming dams was reversed by MET. This finding and the reversal of the downregulation of reelin in offspring of low licking and grooming dams by the HDAC inhibitor trichostatin A suggest that both DNA methylation and histone modifications are involved in the RELN chromatin remodeling attributable to maternal care.
In addition to MET’s ability to downregulate reelin and GAD67 mRNAs and proteins in mice (77), MET added to primary neuronal cell cultures in vitro also downregulates reelin and GAD67 (80). This effect of MET is attenuated by co-transfection of DNMT1 antisense oligonucleotides (80), providing a link between the overexpression of DNMT1 and the reduced expression of reelin and GAD67 in GABAergic neurons found in psychosis and MET-treated mice (32, 42, 45, 49).
6 Pharmacological Correction of Epigenetic GABAergic Downregulation
The objective of developing an epigenetic animal model of GABAergic dysfunction is that once a phenotypic disease model has been induced, the model can be used to explore ways to pharmacologically correct the deficits using drugs that (a) enhance the expression of GABAergic genes reversing chromatin-remodeling abnormalities or (b) act as positive allosteric modulators of GABAA receptors directly to enhance defective GABAergic neurotransmission.
(i)
A possible approach would be to diminish DNA methyltransferase overactivity with inhibitors of DNMTs. However, the most potent DNMT inhibitors (5-azacytidine and zebularine) fail to readily cross the blood–brain barrier when administered systemically and are active only in the S phase of the cell cycle. Hence, there is a search for drugs that could have DNMT inhibitory activity in nondividing differentiated neurons.
(ii)
Nicotinic receptor agonists may have the ability to inhibit DNMT expression in cortical and hippocampal areas. The rate of tobacco smoking in psychiatric patients is very high, especially in SZ patients. Attempts to decrease this behavior have been unsuccessful because withdrawal from nicotine causes an exacerbation of SZ symptoms. Thus, smoking is often viewed as an attempt by patients to self-medicate. Satta et al. (42) administered nicotine to mice and found that nicotine downregulated DNMT1 expression in the frontal cortex and hippocampal areas that express cholinergic nicotinic receptors. Further, the same doses of nicotine that decreased DNMT1 expression also diminished the level of cytosine-5-methylation in the GAD1 promoter and prevented the MET-induced hypermethylation of this promoter (Fig. 11.2). The results suggest that nicotine or other nicotinic agonists, by activating nicotinic cholinergic receptors located on cortical or hippocampal GABAergic interneurons, can upregulate GAD67 expression via modification of an epigenetic mechanism.
(iii)
Another approach to correct abnormal GABAergic promoter methylation is to test drugs that activate DNA demethylation processes. Using the MET mouse model of SZ, we have shown that hypermethylated reelin and GAD1 promoters can be demethylated by the administration of clinically relevant doses of the HDAC inhibitors VPA or MS-275 (6, 81). Because the HDAC inhibitor VPA is frequently used clinically in combination with antipsychotics as an augmentation strategy to treat multiple symptoms, especially cognitive symptoms in patients suffering from BP or schizoaffective disorders, Dong et al. (82) measured the ratio of 5-methylcytosine to unmethylated cytosine in RELN and GAD1 promoters in the mouse frontal cortex and striatum. Normal mice were compared to mice pretreated with MET to hypermethylate these promoters. The results were that clinically relevant doses of clozapine and sulpiride, but not haloperidol or olanzapine, induced dose-related increases in the cortical and striatal demethylation of hypermethylated RELN and GAD67 promoters. The effects of clozapine and sulpiride were dramatically potentiated by a clinically relevant dose of VPA. These findings in mice are consistent with the clinical efficacy in some BP patients of combining VPA with antipsychotic treatment. These data suggest that the induction of DNA demethylation by specific subtypes of antipsychotics or other agents that act at the level of chromatin structure should be further investigated. This may lead to the development of drug treatments effective not only on positive but also on negative symptoms of major psychosis.
(iv)
A further strategy to correct epigenetic GABAergic downregulation in psychosis is to enhance the defective GABAergic transmission using drugs acting as selective positive allosteric modulators of GABA action at pertinent GABA A receptor subtypes (35, 83). Benzodiazepines that are devoid of intrinsic activity at GABAA receptors that express α1 subunits but that act as full positive allosteric modulators of GABA action at GABAA receptors containing α2, α3, or α5 subunits should counteract GABAergic signal transduction deficits without eliciting sedation, amnesia, tolerance, or dependence liability (26). A benzodiazepine with the above-mentioned properties is imidazenil (26). Hence, we suggest that drugs like imidazenil, given alone or in combination with antipsychotics, should be tested in the treatment of GABAergic dysfunction operative in SZ and BP disorder.
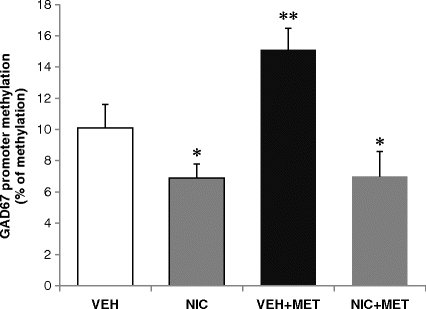
Fig. 11.2.
Nicotine reduces GAD67 promoter methylation in the frontal cortex. VEH, received saline four times a day for 5 days; NIC, received nicotine 3.5 mg/kg s.c. four times a day for 4 days followed by saline on the fifth day; VEH+MET, received saline for 2 days followed by l-methionine (5 mmol/kg, twice a day) for 3 days; NIC+MET, received nicotine (3.5 mg/kg) for 4 days. On the third and fourth days of nicotine treatment, mice also received l-methionine. On the fifth day, nicotine was suspended and mice received only l-methionine. Each value is the mean and ±SE for three mice. One-way ANOVA: Newman–Keuls multiple comparison. ** p < 0.01 VEH + MET vs VEH and VEH + MET vs NIC + MET ; * p < 0.05 VEH vs NIC and VEH vs NIC + MET. (Modified from Satta et al., (42).)
Support for the use of imidazenil in psychoses is provided by experiments with reeler heterozygous mice. In one such experiment (37), imidazenil corrected the deficiency in PPI in heterozygous reeler mice, while alprazolam failed to do so. In addition, administration of imidazenil reduced the PPI and social interaction deficits in mice receiving protracted MET treatment (78). PPI is often viewed as an endophenotype for psychosis vulnerability (84) and it has been shown to reflect α3 and α5 subunit activity (85, 86), which are the subunits of the GABAA receptor affected by imidazenil.
7 Conclusions
This review has focused on epigenetic animal models of SZ or BP disorders characterized by hypermethylation and epigenetic downregulation of forebrain GABAergic genes, even though in the MET-treated mouse model, non-GABAergic promoters can also be hypermethylated. However, interrogating the genome using the ChIP on chip technique indicates that only a relatively small number of genes (<5%) are hypermethylated in the frontal cortex or the hippocampus of mice receiving protracted treatment with MET (82). This selectivity for an epigenetic regulation of a small number of genes has also been reported in rats as a function of variations in maternal care. Offspring show that the vast majority of genes did not change expression (72). Further, normalization by the HDAC inhibitor trichostatin A or methylation by MET involved changes in the expression of a relatively few select gene clusters (72). Hence, these observations would argue against a hypothesis proposing an overall general reprogramming of gene expression in the changes induced by the environment or protracted MET treatment in mice.
A shortcoming of the MET model is that we have not yet found evidence for an upregulation of DNMTs by MET comparable to the elevation of DNMT levels in psychotic postmortem brain, although SAM, which is increased in SZ along with DNMTs, is increased by MET and contributes to the hypermethylation of selective gene promoters (77). In spite of shortcomings, the mouse methionine model continues to be useful, especially in regard to study of the combined use of HDAC inhibitors and antipsychotics. Using this model, we have recently demonstrated that specific antipsychotic subtypes (i.e., clozapine and sulpiride) can cause persistent epigenetic changes by affecting chromatin structure and by inducing GABAergic promoter demethylation (6).
The goal of our studies is to develop a classification of antipsychotic drugs based on their epigenetic action. To achieve this goal, we propose using the MET-treated mouse model to study other typical and atypical antipsychotics to investigate whether antipsychotics that decrease GABAergic promoter methylation act by decreasing DNMTs or by activating a DNA demethylase. We have preliminary evidence suggesting that the molecular mechanisms underpinning GABAergic promoter demethylation caused by clozapine or sulpiride administration may include an activation of a putative DNA demethylation process (82). Because the biochemical identity and function of a putative DNA demethylase in neurons still requires clarification, in the future, its characterization will be crucial for the identification of a new line of pharmacological agents to treat Sz and related major psychiatric disorders.
Acknowledgments
This research was partially supported by NIH grants MH071667 to E. Costa and by MH070855 to A. Guidotti.
References
1.
2.
3.
Ptak, C., Petronis, A. (2008) Epigenetics and complex disease: from etiology to new therapeutics. Ann. Rev. Parmacol. Toxicol. 48, 257–276.CrossRef
4.
5.
6.
7.
8.
Epsztejn-Litman, S., Feldman, N., Abu-Ramaileh, M., Shufaro, Y., Gerson, A., Ueda, J., Depius, R., Fuks, F., Shinkai, Y., Cedar, H., Bergman, Y. (2008) De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15, 1176–1183.PubMedCrossRef
9.
10.
11.
Szyf, M. (2009) The early life environment and the epigenome. Biochim. Biophys. Acta 1790, 878–885.PubMed
12.
Ooi, S.K.T., Bestor, T.H. (2008) The colorful history of active DNA demethylation. Cell 133, 145–148.CrossRef
13.
14.
15.
16.
Grayson, D.R., Chen, Y., Costa, E., Dong, E., Guidotti, A., Kundakovic, M., Sharma, R.P. (2006) The human reelin gene: transcription factors (+), repressors (–) and the methylation switch (+/1) in schizophrenia. Pharm. Therap. 111, 272–286.CrossRef
17.
Huang, H.-S., Akbarian, S. (2007) GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PloS One 8, e809.CrossRef
18.
Fatemi, S.H., Stary, J.M., Earle, J.A., Araghi-Nikham, M., Egan, E. (2005) GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and reelin proteins in cerebellum. Schizophr. Res. 72, 109–122.PubMedCrossRef
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


