CHAPTER 37 Endotoxemia
ETIOLOGY
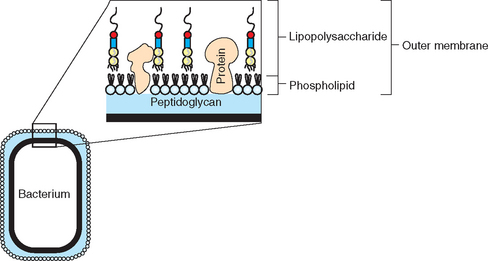
The clinical syndrome of endotoxemia is the result of a massive, dysregulated, and generalized inflammatory response to endotoxin, the major structural component of the outer membrane layer of the gram-negative bacterial cell wall (Fig. 37-1). Sources of endotoxin relevant to equine medicine include gram-negative pathogens, as well as gram-negative bacteria that are part of the normal intestinal, especially cecal, microflora. Endotoxemia associated with gram-negative infections may result from generalized disease, such as neonatal septicemia, or from more localized processes, such as pleuropneumonia, peritonitis, or endometritis. With localized infections, “spillover” of inflammatory mediators into the systemic circulation may occur once a critical concentration has been reached at the local site.1
Translocation of endotoxin from the intestinal lumen may occur on compromise of the mucosal barrier, which is most pronounced with severe inflammation, such as during acute colitis, or with ischemia, as in strangulating intestinal lesions. Other suggested causes of endotoxin translocation include severe shock states, severe trauma, malnutrition, and strenuous exercise.2–6 Mechanisms of gut barrier compromise under these conditions may include reduced intestinal blood flow, resulting in ischemia; hypoxemia; and increased body temperature. The relevance of endotoxin translocation from these causes has not been investigated sufficiently in horses; however, these may serve to explain the occurrence of endotoxemia in compromised patients without primary infection or intestinal disease.
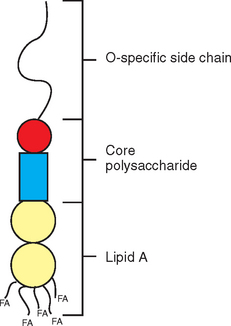
Endotoxin molecules have lipopolysaccharide (LPS) structure and consist of three distinct portions: the O-polysaccharide (or O-chain), core oligosaccharide (divided further into inner and outer core), and lipid A (Fig. 37-2). Lipid A represents the biologically active portion of LPS and anchors the endotoxin molecule in the cell membrane; therefore it is not available for interaction with inflammatory cells until endotoxin is released from the bacterial cell.7 This release occurs on cell division and cell death, but also on bacterial killing from antimicrobial treatment.8 After release from the bacterial cell, endotoxin molecules tend to aggregate and form micelles based on their amphipathic nature. In plasma, individual LPS molecules can be removed from these micellar aggregates by plasma proteins, most notably lipopolysaccharide-binding protein (LBP).9 LBP then transfers LPS monomers to the surface of inflammatory cells, where the molecules bind to a specific receptor complex and elicit cell activation. Increased production and release of cytokines and other inflammatory mediators in response to this cell activation represents a crucial step in initiation and maintenance of the inflammatory cascade.
The structure of endotoxin, particularly the lipid A portion, is well conserved among pathogenic gram-negative bacteria. Research in recent years has shown that a group of “pathogen-associated molecular patterns” (PAMPs) play the role of “recognition signals” for the presence of pathogens. These PAMPs are molecules that are consistently present in certain types of pathogens and are structurally conserved. Rather than nonspecifically stimulating immune cells, PAMPs are recognized by “hard-wired”10 receptors of the innate immune system, which as a group are referred to as “pattern recognition receptors” (PRRs). Endotoxin can be regarded as the prototypic PAMP signaling the presence of gram-negative bacteria, and toll-like receptor 4 (TLR4) has been identified as the PRR for endotoxin.11 In addition to TLR4, 10 other TLRs have been identified to date, and most of their ligand specificities have been identified (Table 37-1). TLR4 functions as part of a receptor complex that also includes the cluster of differentiation antigen 14 (CD14) and myeloid differentiation factor-2 (MD-2), a small protein that interacts with the extracellular portion of TLR4. Although all three proteins appear to be required to provide a high sensitivity of cells to endotoxin, TLR4 is the only protein with a transmembrane domain and therefore deserves particular recognition when investigating cellular stimulation by endotoxin.
Table 37-1 Toll-Like Receptor (TLR) Ligands
| RECEPTOR | MICROBIAL AND SYNTHETIC LIGANDS |
|---|---|
| TLR1 | Triacyl lipopeptides (e.g., Pam3Cys) |
| TLR2 | Lipoproteins, lipopeptides, peptidoglycan, yeast zymosan, lipoteichoic acid, atypical lipopolysaccharide (LPS), lipoarabinomannan |
| TLR3 | Double-stranded RNA |
| TLR4 | LPS, taxol, viral F-protein (e.g., respiratory syncytial virus) |
| TLR5 | Flagellin |
| TLR6 | Diacyl lipopeptides, yeast zymosan, lipoteichoic acid |
| TLR7 | Imidazoquinoline, loxoribine, single-stranded RNA viruses |
| TLR8 | Imidazoquinoline, single-stranded RNA viruses |
| TLR9 | CpG DNA, CpG oligonucleotides |
| TLR10 | Not identified |
| TLR11 | Ligands derived from uropathogenic bacteria |
Modified from Horner AA, Redecke V, Raz E: Curr Opin All Clin Immunol 4:6,555-561, 2004.
Similarities of the inflammatory response extend to other inciting causes, such as gram-positive bacterial, viral, or fungal infection and severe trauma. The term systemic inflammatory response syndrome (SIRS)12 was introduced to describe the clinical entity of an overzealous inflammatory response in the absence of an etiologic diagnosis. For the purpose of case definition in human clinical trials, SIRS is defined by the presence of at least two characteristic findings, including hypothermia or hyperthermia; tachycardia, tachypnea, or hypocapnia; and leukocytosis, leukopenia, or an increased number of immature leukocyte forms.12 These criteria certainly apply to many horses currently diagnosed as being “endotoxemic,” and therefore use of the term SIRS in equine medicine may be appropriate and potentially more useful.13
Additional definitions applicable to human patients include sepsis (SIRS caused by demonstrable infection, whether localized or in the form of bacteremia), severe sepsis, and septic shock (sepsis-induced hypotension, persisting despite adequate fluid resuscitation, along with the presence of hypoperfusion abnormalities or organ dysfunction).12 Multiple organ dysfunction syndrome is defined as insufficiency of two or more organs, which may manifest as clinical changes or may be diagnosed based on clinicopathologic data. An adjustment of this terminology to the equine patient has not been undertaken to date but might offer advantages with respect to case definition for clinical trials as well as establishment of prognostic criteria for “endotoxemic” patients.
EPIDEMIOLOGY
Endotoxemia affects horses and ponies of either gender and of all breeds and ages, including neonatal foals. Studies performed at referral institutions have found that 10% to 40% of horses presented for colic14–16 and 50% of septic neonatal foals17 had measurable circulating endotoxin concentrations. The number of colic patients testing positive for endotoxin was increased when only horses with conditions requiring exploratory surgery were investigated.
Individual differences in the sensitivity to endotoxin, although anecdotally observed, have not been documented in horses. In other species, including humans, presence of certain gene polymorphisms has been associated with a reduced sensitivity to endotoxin challenge (“hyporesponders”) and may also be associated with disease susceptibility and outcome. Gene polymorphisms are defined as allelic variants that exist stably in a population and occur in frequencies (>1%) not attributable to new mutations.18 Polymorphisms most often occur as single-nucleotide polymorphisms (SNPs) and may affect coding as well as noncoding regions of genes.
SNPs within coding regions (exons) that result in altered amino acid sequence presumably affect protein function. For example, SNPs in the coding region of the human TLR4 gene are associated with impaired signal transduction in response to LPS stimulation.19 An association between SNPs in human TLR4 and unfavorable outcome of sepsis in human patients has been suggested20,21 and may be attributable to a reduced immune defense against infection favoring the development of sepsis. Changes in noncoding regions (introns) or in gene promoters may alter transcription rate and messenger ribonucleic acid (mRNA) stability or may serve as “markers” for certain traits if they are in linkage with biologically active SNPs in other gene regions.18
A polymorphism in the human tumor necrosis factor alpha (TNF-α) gene promoter22 may affect the magnitude of TNF-α expression on cell stimulation and is likely associated with outcome of septic shock. Polymorphisms in promoter regions of human interleukin-6 (IL-6) and interleukin-10 (IL-10) are associated with decreased cytokine production in vitro, although their clinical relevance has not been evaluated to date.18 The biologic significance of SNPs in the equine TNF-α gene promoter23 has not been determined. Investigation of gene polymorphisms currently is an area of great interest as it may provide the basis for a more individualized approach to patient assessment and therapy.
PATHOGENESIS
Inflammatory Cell Activation
Investigation is ongoing as to the exact mechanism by which endotoxin binds to its cellular receptor complex and initiates cell activation, as well as the details of intracellular signaling in response to cell stimulation. One obstacle in the evaluation of ligand-receptor interactions is the current lack of understanding of the three-dimensional structure of LPS receptor proteins and therefore the LPS-binding site(s). As mentioned earlier, at least three proteins (CD14, TLR4, MD-2) constitute the major receptor complex for LPS, and interaction of LPS with all proteins during initiation of cell signaling has been demonstrated experimentally.24 Additional binding proteins, which may be involved in the initiation of cell signaling and cell activation, include β2-integrins, moesin, heat shock proteins, chemokine receptors, growth differentiation factors, and intracellular nucleotide-binding oligomerization domain (NOD)1 and NOD2.7,25–27
CD14 was the first protein known to be essential for LPS binding and cellular activation.28 CD14 is a 60-kilodalton (kDa) protein that is constitutively expressed on myeloid cells, including monocytes, macrophages, and neutrophils, as well as B lymphocytes and several other cell types.7 In horses experimentally infused with endotoxin, an increase in the number of circulating CD14-expressing cells, as well as in the number of immunoglobulin M (IgM)–positive and major histocompatibility complex (MHC) class II–positive cells, correlated with changes in vital parameters.29 In addition to membrane-bound forms (mCD14), a soluble form (sCD14) is present in plasma and may be able to transfer LPS to non–CD14-bearing cells (e.g., endothelial cells) to render these cells endotoxin responsive.30 Conversely, high concentrations of sCD14 may have “detoxifying” effects by scavenging LPS and preventing it from interacting with inflammatory cells. This “bipolar” effect has similarly been suggested for LBP.
TLR4 in association with MD-2 represents the actual signaling portion of the LPS receptor complex.11,31 TLR4 is a 92-kDa type I transmembrane receptor with an interleukin-1 receptor (IL-1R)–like intracellular domain. Similarities to IL-1R extend beyond structure, and signaling pathways in response to TLR4 stimulation or IL-1R stimulation show substantial similarity. TLR4 is expressed constitutively on neutrophils, monocytes, macrophages, and dendritic cells, but also on epithelial and endothelial cells. Expression levels vary among individuals and are regulated in a tissue-specific manner by LPS and cytokines (e.g., TNF-α). Oligomerization is required for signaling through TLRs, and MD-2 may have an effect on TLR4 aggregation on the cell surface.32 MD-2 is a small, secreted glycoprotein that associates with the extracellular domain of TLR4 and is most likely required for proper receptor function. MD-2 may also be involved in ensuring proper glycosylation and transport of TLR4 to the cellular surface.
An interesting observation is that certain “atypical” LPS molecules, which show variation from enteric LPS in their lipid A structure, can act as endotoxin antagonists. These molecules compete for receptor binding but do not cause cell activation, and in effect they block cellular response to “agonist” endotoxin.33 Curiously, recognition of LPS molecules as agonists or antagonists depends on the host species, which has prompted investigations into the determinants of receptor specificity. Uniformly, these studies have shown that the species origin of CD14 does not determine receptor specificity, a finding that further supports a primary role for CD14 as an endotoxin capture molecule. Conclusions concerning the relative role of TLR4 and MD-2 have been variable; some studies suggest that either TLR4 or MD-2 is the primary determinant of receptor specificity, and others suggest that both proteins play a role.34–36 Cell transfection experiments performed in vitro suggest that the complex of TLR4 and MD-2 mediates recognition of endotoxic LPS in equine cells.37
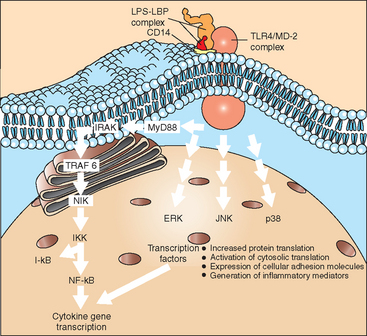
Intracellular signaling pathways resulting in activation of nuclear factor kappa B (NF-κB) and stimulation of mitogen-activated protein (MAP) kinases are of major significance for cell activation by LPS (Fig. 37-3), although additional pathways have been suggested. In effect, cell-signaling events culminate in the translocation of transcription factors to the cell nucleus, with a resulting increase in gene transcription and protein expression. Translational and posttranslational processes may also be activated, such that the regulation of protein expression in response to endotoxin is a complex event offering multiple targets for regulation and potential therapeutic influence.
Inflammatory Mediators
The pathogenesis of endotoxemia is primarily determined by the effects of inflammatory mediators, many of which are released after inflammatory cell stimulation. Important mediators include the cytokines, prostaglandins, thromboxanes, leukotrienes, platelet-activating factor, nitric oxide, reactive oxygen species, histamine, kinins, complement components, and growth factors. Table 37-2 summarizes the origin, regulation, and biologic effects of some of the major inflammatory mediators. Cytokines play a particularly important role because they not only exert pathophysiologically relevant effects, but also regulate the release of other mediators. Primarily, proinflammatory cytokines (e.g., TNF-α, IL-1) are central to the initiation and maintenance of an inflammatory reaction, and development of SIRS has classically been attributed to an uncontrolled overproduction of these proinflammatory mediators.1
Table 37-2 Important Mediators of the Systemic Inflammatory Response to Endotoxin
| MEDIATOR | ORIGIN | EFFECTS |
|---|---|---|
| Tumor necrosis factor (TNF) | ||
| Interleukin-1 (IL-1) | ||
| Interleukin-6 (IL-6) | ||
| Interleukin-8 (IL-8) | Neutrophil activation and chemotaxis | |
| Interleukin-10 (IL-10) | ||
| Thromboxane A2 (TxA2) | Platelets | |
| Prostaglandin E2 (PGE2) | Most nucleated cells | |
| Prostaglandin I2 (PGI2) | Vascular endothelial cells | |
| Prostaglandin F2α (PGF2α) | Most nucleated cells | |
| Platelet-activating factor (PAF) | ||
| Leukotriene B4 (LTB4) | ||
| Leukotrienes C4, D4, E4 (SRS-A) | ||
| Kinins | Produced from serum precursors | |
| Complement components (C3a, C5a) | — | |
| Oxygen-derived free radicals | ||
| Granulocyte-monocyte colony-stimulating factor (GM-CSF) | — | Rebound neutrophilia |
SRS-A, Slow-reacting substance of anaphylaxis.
In recent years, however, studies have increasingly investigated cytokines with mostly antiinflammatory activity, such as IL-10, IL-4, IL-11, and transforming growth factor beta (TGF-β), which are produced along with their proinflammatory counterparts and may serve to contain and attenuate inflammation after an invading pathogen has been eliminated. In addition to these cytokines, downregulating effects on the inflammatory cascade are attributed to soluble cytokine receptors (e.g., soluble TNF receptor) and cytokine receptor antagonists (e.g., soluble IL-1R antagonist), which inhibit cytokine effects at the level of the inflammatory cell. The role of antiinflammatory responses in disease development is not completely understood. However, a compensatory antiinflammatory response syndrome (CARS) has been described in which excessive production of antiinflammatory mediators results in immunosuppression and ultimately immune anergy.38
The paradigm of “immunologic dissonance,” as proposed by Bone,1 therefore provides a more comprehensive and flexible view of inflammatory responses during infection and endotoxemia. According to this paradigm, every severe insult produces a response consisting of both proinflammatory and antiinflammatory components, and it is the relative balance (or lack thereof) of these components that determines outcome in the form of reestablishment of homeostasis or disease progression toward shock.
Some of the “earliest” or most “proximal” cytokines produced in response to endotoxin include TNF-α, IL-1, and IL-6. The importance of TNF-α in particular is underscored by experiments showing that TNF-α has very similar effects to endotoxin when administered experimentally,39 and that blockade of TNF-α during early endotoxemia abrogates the development of clinical disease in laboratory animals40 and horses.41 In septic human patients, the magnitude of increase in TNF-α and IL-6 concentration is correlated with the severity of sepsis, whereas concentrations of IL-1 are often undetectable and show poor correlation with disease severity.42 In horses, plasma activity of TNF-α is positively correlated with mortality in patients with acute gastrointestinal disease and in septic neonates.14,43,44
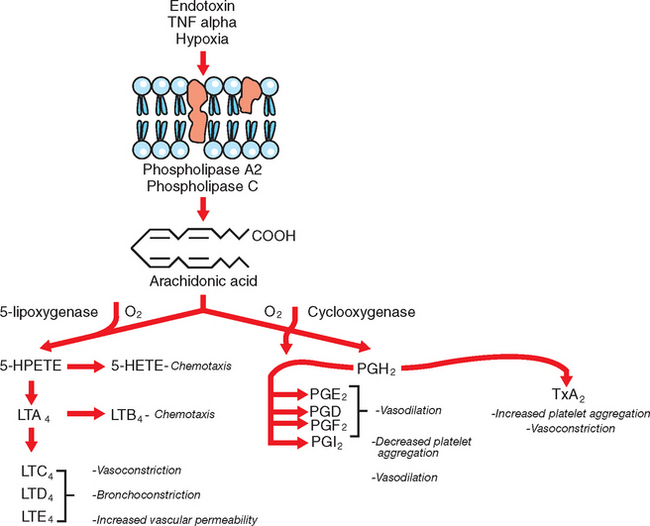
Eicosanoids are arachodonic acid (AA) metabolites and include the prostaglandins, thromboxanes, leukotrienes, lipoxins, hydroxyeicosatetraenoic acid (HETE), and epoxides.45 The rate-limiting step in the production of eicosanoids is the liberation of AA from phospholipid pools in the cell membrane, followed by metabolism of AA through several different pathways (Fig. 37-4). The most important eicosanoids in the pathophysiology of endotoxemia are thromboxane A2 (TxA2) and the prostaglandins E2 (PGE2), I2 (PGI2), and F2α (PGF2α); the significance of leukotrienes is less well understood.46 TxA2 promotes platelet aggregation, vasoconstriction, bronchoconstriction, and leukocyte adhesion;47 PGE2 and particularly PGI2 (prostacyclin) cause vasodilation and have platelet antiaggregating effects. PGE2 and PGI2 also exert antiinflammatory effects by reducing cytokine production, inhibiting activation and proliferation of B cells, decreasing macrophage phagocytosis, and inhibiting neutrophil functions.45 In horses, increased plasma concentration of TxA2 has been associated with very early responses to experimental endotoxemia, including pulmonary hypertension, dyspnea, and hypoxemia, whereas a rise in PGI2 occurred concurrently with clinical signs of abdominal pain, mucous membrane discoloration, prolongation of the capillary refill time, and the development of hypotension.48
Endothelial Dysfunction, Hemodynamic Changes, and Shock
Endotoxic shock is generally classified as “distributive shock” and is largely attributable to peripheral vascular dysfunction resulting in maldistribution of blood flow and perfusion deficits. Endotoxin exerts peripheral vasomotor effects through release of inflammatory mediators such as prostacyclin and nitric oxide, which cause widespread vasodilation and vasoplegia, leading to blood pooling in the periphery and a reduction of effective circulating volume.49 In addition, cardiac function is compromised by decreased coronary blood flow and the release of myocardial depressant factors; circulating volume is reduced by increased vascular permeability; and tissue oxygen extraction is impaired. Initially, tachycardia and increased cardiac output as well as increases in central venous pressure and pulmonary arterial pressure (hyperdynamic phase) may have a compensatory effect.49 However, disease progression is characterized by the development of systemic hypotension and ultimately perfusion deficits of vital organs.
Neutrophil Activation
Neutrophil activation during bacterial infection generally serves to promote extravasation into infected tissues and increase the cells’ bactericidal capacity. Activated neutrophils express adhesion molecules for interaction with endothelial cells, exhibit an increased capacity for phagocytosis and respiratory burst, and release lysosomal enzymes and inflammatory mediators.50 Neutrophil margination and extravasation occur in three phases, characterized by the expression of different adhesion molecules on neutrophils and endothelial cells. The first phase of neutrophil tethering and rolling is mediated by P-selectin and E-selectin on endothelial cells, which interact with P-selectin glycoprotein ligand-1 (PSGL-1) and sialylated Lewis-X–like structures on leukocytes. Firm adhesion of neutrophils to the endothelium during the second phase results from the interaction between endothelial intercellular adhesion molecule-1 (ICAM-1) and neutrophil integrins leukocyte function–associated antigen-1 (LFA-1) and Mac-1. The third phase of neutrophil transmigration depends on expression of platelet/endothelial cell adhesion molecule-1, located at the intercellular junction of endothelial cells.
Neutropenia is an early finding during experimental endotoxin administration and may be the only specific clinicopathologic evidence of acute sepsis or endotoxemia.51 Neutrophil margination is observed particularly within the lung vasculature,52 and neutrophils are important players in the development of acute lung injury after endotoxin administration. Mechanisms of neutrophil-mediated lung injury may include vascular endothelial damage, as outlined in the previous section; neutrophil migration into airways; expression of proinflammatory cytokines (e.g., IL-1, TNF-α); and oxidant-induced injury resulting in loss of epithelial integrity.53 In some cases, these events may culminate in the development of a “shock lung,”54 and pulmonary failure remains the leading cause of sepsis-related death in human patients.
Recent investigations using chimeric mouse models have shown that activation of circulating neutrophils by endotoxin is not sufficient to induce neutrophil sequestration within the lung. These studies have suggested that TLR4-mediated endothelial cell activation is the dominant event in the development of inappropriate leukocyte trafficking during endotoxemia.55 On recovery from endotoxemia or infection, reentry of marginated neutrophils into the circulation leads to rebound neutrophilia; however, it is questionable whether these neutrophils exhibit normal cellular function and signify a recovery of normal immune responses. Increased release of neutrophils from the bone marrow caused by stimulation of myeloid cell proliferation by granulocyte-macrophage colony-stimulating factor (GM-CSF) also contributes to rebound neutrophilia.
Coagulopathy
Coagulopathy often develops during endotoxemia and has been described in horses with colic,56–58 as well as in septic foals.17 Coagulopathy develops because of simultaneous activation of coagulation and fibrinolysis, and in its most severe form, results in consumptive coagulopathy and disseminated intravascular coagulation (DIC).59 Usually, however, clinical signs of coagulopathy are limited to an increased thrombotic tendency (e.g., jugular venous thrombosis) or an increased bleeding tendency (e.g., following venipuncture or nasogastric intubation). Diffuse microthrombosis may contribute to tissue ischemia and the development of organ failure. The factors that determine whether activation of procoagulant or fibrinolytic pathways predominates are poorly understood. Because an abnormal coagulation profile can be detected before the development of clinical signs of coagulopathy, evaluation of coagulation parameters is an important step in patient assessment, and abnormalities should prompt preemptive treatment to prevent further deterioration.
Activation of coagulation is by both the intrinsic and the extrinsic pathway and culminates in thrombin-mediated conversion of fibrinogen into fibrin. Endotoxin directly activates the intrinsic coagulation pathway through the contact system comprising factor XII (Hageman factor), prekallikrein, and high-molecular-weight kininogen. More importantly, however, activation of the extrinsic pathway results from exposure of subendothelial tissue factor after endothelial damage and stimulation of tissue factor expression on activated mononuclear phagocytes and endothelial cells. Increased expression of monocyte tissue factor (also referred to as “procoagulant activity”) is reported to be significantly associated with poor prognosis in equine colic patients.60 Tissue factor expression by peritoneal macrophages may favor the development of intraabdominal adhesions in colic patients undergoing exploratory laparotomy.61 Tissue factor forms complexes with coagulation factor VIIa and subsequently activates factors X and IX. Factor Xa associates with factor Va to effect prothrombin conversion into thrombin (prothrombinase complex), whereas factor IXa in association with factor VIIIa further activates factor X activation. Thrombin is formed initially on tissue factor–bearing surfaces and cells and subsequently stimulates amplified thrombin production on the platelet surface.62 Thrombin stimulates platelet adhesion and activation and activates clotting factors V, VIII, and XI. In addition, platelet aggregation is favored by increased release of TxA2 from activated vascular endothelial cells and release of TxA2 and platelet-activating factor (PAF) from activated platelets. A predominant role for PAF rather than TxA2 in endotoxin-induced platelet aggregation has been suggested.63,64
Procoagulant tendency during endotoxemia is further attributable to impairment of regulatory (i.e., anticoagulant) mechanisms. For example, endothelial dysfunction leads to decreased activation of protein C as well as decreased expression of antithrombin III. Activated protein C (APC) is important for the anticoagulant properties of normal endothelium and acts in concert with protein S by inactivating clotting factors Va and VIIIa as well as plasminogen activator inhibitor (PAI).65 Production of APC depends on thrombin interaction with thrombomodulin on the vascular endothelium, such that decreased thrombomodulin expression by damaged endothelial cells decreases APC formation. Antithrombin III inhibits multiple components of both the intrinsic and the extrinsic coagulation pathway, including clotting factors IIa, IXa, Xa, XIa, XIIa, VIIa/tissue factor, and kallikrein.66
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


