Chapter 20 Drugs Affecting the Respiratory System
Normal Respiratory Physiology
Airway Caliber Changes
Nervous innervation to the smooth muscle of the respiratory tract is complex. The parasympathetic system provides the primary efferent innervation, with acetylcholine as the primary neurotransmitter.1,2 Its fibers are responsible for the baseline tone of mild bronchoconstriction that characterizes the normal respiratory tract through M3 muscarinic receptors. The effects are balanced by bronchodilation resulting from β2-receptor stimulation.3–5, In contrast, α-adrenergic stimulation can contribute to bronchoconstriction.2,4,6 A third, largely understood nervous system, referred to as the nonadrenergic, noncholinergic system or the purinergic system, also innervates bronchial smooth muscle.1,7 This system mediates bronchodilation by way of vagal stimulation. The afferent fibers of this system are probably irritant receptors, and although the neurotransmitter has not yet been conclusively identified, vasoactive intestinal peptide has been implicated in the cat.8,9 Malfunction of this system has been associated with bronchial hyperreactivity, which often characterizes asthma.7
The intracellular mechanisms that transmit signals from the nervous system to smooth muscle depend in part on changes in the intracellular concentration of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) (Figure 20-1). The effects of these two secondary messengers are reciprocal such that the increased intracellular concentration of one is associated with a decreased concentration of the other. Cyclic AMP-induced bronchodilation is decreased by α-adrenergic stimulation and increased by β2-receptor stimulation.3 In contrast, cGMP-induced bronchoconstriction is increased by stimulation of muscarinic (cholinergic) and, indirectly, histaminergic receptors (see Figure 20-1). The relative sensitivity of bronchial smooth muscle to histamine-induced and acetylcholine-induced bronchoconstriction varies with the location and species.5,10,11 Peripheral airways in dogs are more susceptible than in cats to acetylcholine; feline airways, in general, are more sensitive to acetylcholine and serotonin than histamine.12 Smooth muscle receptors are also susceptible to stimulation by a variety of chemical mediators (see Figure 20-1), which may also modulate cAMP and cGMP.13–15
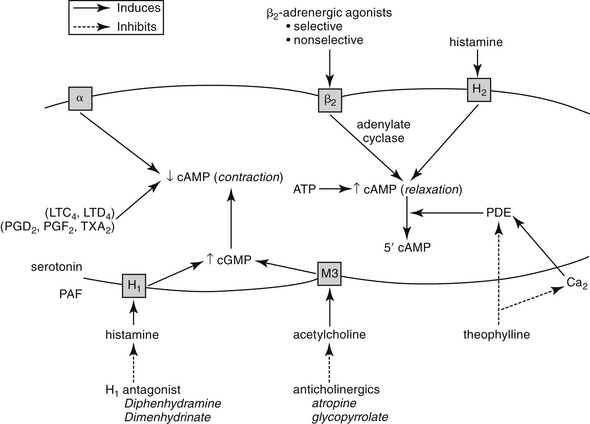
Control of bronchial smooth muscle tone is very complex and depends on input from sensory receptors. At least five types of sensory receptors have been identified in cat lungs, all of which can be classified as irritant (or mechanoreceptor), stretch, or J-receptors.7 All appear to be innervated by the parasympathetic system. Irritant receptors, located beneath the respiratory epithelium, occur in the upper airways2 and, in cats, as far peripherally as the alveoli.1 Physical, mechanical, or chemical stimulation of these receptors results in tachypnea, bronchoconstriction, and cough. As reviewed by Canning,7a rapidly adapting receptors (RARs) terminate in or below the epithelium primarily of intra-pulmonary, but to some degree, extra-pulmonary airways. They can adapt within 1 to 2 seconds to changes in airway mechanics and are subject to activation by multiple stimuli, including chemical mediators. In contrast, slowly adapting receptors (SARs) may be responsible for physiologic responses (i.e., termination of inspiration and initiation of expiration). SARS centrally inhibit respiration and decrease airway smooth muscle tone. However, they may facilitate cough. Cats have few SARS but many RARs.7a C-fibers represent the majority of fibers innervating the airways and lungs. They are physiologically similar to nociceptors of other tissues. They appear to synthesize neuropeptides (e.g., substance P) that influence the central and peripheral nerve terminals. Mediators stimulating C-fibers vary: in dogs, pulmonary subtypes do not (but bronchial subtypes do) respond to histamine, and in other animal models, they do not respond to serotonin or adenosine. C-fibers regulate airway defense reflexes, interacting with RAR, although the scientific description of these fibers is not clear. C-fibers and RARS interact with the spinal cord through central sensitization. Subsequent heightened reflex responsiveness may occur; tachykinins (including substance P) appear to play a role in hyperreflexia, an effect prevented by neurokinin receptor antagonists. Convergence of vagal afferents in the nucleus of the solitary tract may explain in part the close relationship between vomiting and coughing. Receptors also converge in the ventral respiratory column of the medulla. Airflow velocity appears to be the most critical factor determining stimulation of irritant receptors in the upper airways.1 Airway constriction sufficient to cause airflow velocity to exceed a specific threshold results in a vagally mediated cough reflex and bronchoconstriction. Airways can also be occluded by mucus and edema or by chemical mediators released during upper airway infections.7
Respiratory Defense Mechanisms
In addition to the cough and sneeze reflexes, two other systems provide the major defense of the respiratory tract against invading organisms or foreign materials: the mucociliary apparatus and the respiratory mononuclear phagocyte system.2 The mucociliary apparatus (Figure 20-2) is the first major defense and consists of the ciliary lining of the tracheobronchial tree and the fluid blanket surrounding the cilia. Nervous innervation to the cilia has not yet been identified. Although ciliary activity increases with β-adrenergic stimulation, this may simply reflect the sequelae of β-adrenergic stimulation on respiratory secretions.16 Two types of secretions form the fluid blanket of the respiratory tract.17 The cilia must be surrounded by a low-viscosity, watery medium to maintain their rhythmic beat. A more mucoid layer lies on top of the cilia and serves to trap foreign materials inspired with air. The synchronous motion of the cilia causes the cephalad movement of the mucous layer and any trapped materials.
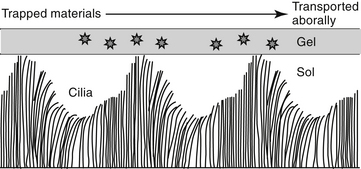
Changes in the viscoelastic properties of mucus such that it becomes either too watery or too rigid will result in mucus transport that is less than optimal.2 Mucus released by goblet cells results from direct irritation2 and is not amenable to pharmacologic manipulation. Surface goblet cells, which are uniquely prominent in feline bronchioles,18 increase in number with chronic disease. Submucosal glands of the bronchi secrete both a serous and a mucoid fluid. The secretions tend to be more fluid than that of the goblet cells, but the degree varies with the stimulus. The normal consistency of the combined secretions of the tracheobronchial tree is 95% water, 2% glycoprotein, 1% carbohydrate, and less than 1% lipid.2 Glycoproteins increase the viscosity of the secretions, providing protection and lubrication. Infection and chronic inflammatory diseases can have a profound effect on respiratory secretions. The glycoprotein component tends to be replaced by degradative products of inflammation such as DNA and actin.17 Goblet cell numbers increase with a subsequent increase in the viscosity of respiration secretion. Parasympathetic, cholinergic stimulation increases mucus secretion, whereas β-adrenergic stimulation causes secretion of mucus, electrolytes, and water.2,16
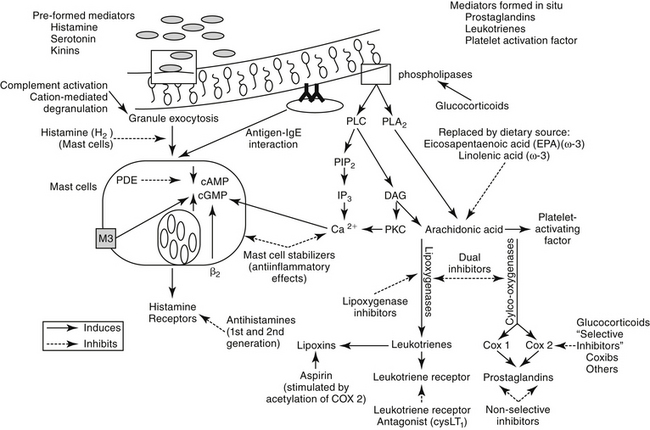
The second major component of the pulmonary defense system is the respiratory mononuclear phagocyte system. In cats, calves, pigs, sheep, and goats, this includes both alveolar macrophages and the pulmonary intravascular macrophages (PIMs).19 The PIMs are resident cells that are characterized by phagocytic properties and thus cause the release of inflammatory mediators. The clearance of blood-borne bacteria and particulate matter in these species is accomplished by PIMs rather than hepatic Kupffer cells and splenic macrophages as in most other species.19 The pharmacologic significance of the mononuclear phagocyte system reflects their role in inflammation (Figure 20-3; see also Figure 20-1). A number of preformed (e.g., histamine and serotonin) and in situ (e.g., prostaglandins, leukotrienes, and platelet-activating factor [PAF]) mediators are released by inflammation cells.13–15 Each is capable of inducing a variety of adverse effects that tend to decrease airway caliber size: edema, chemotaxis, increased mucus production, and bronchoconstriction (Table 20-1). The involvement of PIMs in both experimental and natural respiratory diseases of animals suggests that release of chemical mediators from these cells may be important in the pathogenesis of bronchial diseases.
Surfactant
The primary function of surfactant is to decrease surface tension in alveoli, affecting lung mechanics and gas exchange. However, pulmonary surfactant also affects pulmonary defense mechanisms.20 Surfactant is a complex mixture of lipids and proteins synthesized and secreted by alveolar type II cells with relative consistency among mammalian species.20 In humans the proportion of components is predominantly phospholipids (80%) but includes proteins (12%) and other lipids (8%). The phospolipids principally comprise phosphatidylcholine (85%), the most important to reduction of surface tension being dipalmitoylated phosphatidylcholine (DPPC). Four surfactant-associated proteins (SP) are named A through D. Both SP-A and SP-D enhance phagycytosis of bacteria and viruses and regulate type II pneumocytes. The primary role of SP-B and SP-C is reduction of surface tension, particularly in terminal airways. A number of nonspecific host defense mechanisms have been attributed to surfactant.20 Enhanced stability of the alveolar lining film allows it to serve as a nonspecific barrier to microorganism adhesion and subsequent pulmonary invasion. Muciliary transport is improved by its viscoelastic and rheologic properties. Particle clearance is enhanced, in part through an apparently stimulatory effect on chloride ion transport. Antiinflammatory effects reflect superoxide dismutase– and catalase-mediated oxygen radical scavenging and reduced neutrophil production of superoxide.20,21 Direct antibacterial and antiviral properties have also been attributed to surfactant. Both SP-A and SP-D appear to provide a first line of defense by acting as collectins that target carbohydrate structures on a variety of pathogens. Alveolar macrophage response is increased by SP-A; SP-D enhances macrophage release of oxygen radicals. Cytokines (e.g., tumor necrosis factor [TNF]-α interleukin [IL]-1β, and IL-6) release is stimulated by SP-A.
Changes in surfactant have been associated with a number of respiratory diseases. In humans acute respiratory distress syndrome (ARDS) is associated with changes in the biochemical and biophysical characteristics of surfactant. As a result, surface tension increases, and the phospholipid, fatty acid, and protein profiles change. Serum proteins and inflammatory mediators directly inhibit or degrade surfactant. Abnormalities in the amount and both the phospholipid and the protein composition of surfactant have been associated with infectious diseases of the lungs. The ability of surfactant to suppress immune-mediated lung injury has been interpreted as a possible role of surfactant dysfunction in asthma. Elastase-induced dysfunction and impaired SP-A and -D also may contribute to the surface destruction associated with chronic obstructive pulmonary disease (COPD) and emphysema.20
Pathogenesis of Inflammatory Respiratory Diseases
Asthma is a pathologic state of the lungs characterized by marked bronchoconstriction and inflammation.22–26 Although recent evidence is emerging that airway smooth muscle plays a role in asthma independent of inflammation,27 the latter is an important contributor to disease and warrants a continued focus for drug therapy. Airways become hypersensitive to selected mediators (e.g., histamine and cholinergic stimulants).5,10 The terms allergy and atopy are often used interchangeably; however, for this discussion allergy refers to an uncommitted biological response to an antigen. The pathophysiology of chronic allergic diseases is discussed in Chapter 31. Atopy refers to an allergic response characterized by immunoglobulin E (IgE)–mediated antibodies. Atopy generally reflects a genetic predisposition to IgE antibody production against environmental allergens. Most commonly, the allergic response is low grade and thus beneficial (removal of inciting allergen). However, in atopic patients the response, including IgE antibody production, is exaggerated. Asthma is one of several chronic atopic diseases characterized by an increasing incidence in human medicine; others include allergic rhinitis, atopic eczema, and inflammatory bowel disease.28 Inflammatory mediators are the major contributors of the pathophysiology in asthma (see Table 20-1).22,23,25,26,29,30 Studies in several species have shown that initial exposure to an allergen activate mast cells, macrophages, and other cells lining the airways, increasing mucosal epithelial permeability. Subsequent mediator release not only directly and intensely affects airway inflammation, contraction of smooth muscle, and capillary permeability but also initiates release of chemotactic factors, local infiltration of activated eosinophils, and activation of T cells.31
The critical role of T cells in the initiation and perpetuation of asthma has been well documented32 and is supported by response in asthmatics to both glucocorticoids and cyclosporine.32,33 The exaggerated response that characterizes the atopic patient appears to reflect an imbalance in Th1 versus Th2 helper cells. Researchers in human medicine have postulated that atopic individuals fail (possibly as infants) to transition from a Th2-primary response (mediated by ILs 4, 5, and 13) to an “allergy protective” Th1-mediated primary response. The Th1 response is dominated by cell-mediated immunity, and delayed hypersensitivity (cytotoxic T cells) is initiated through macrophage production of IL-12 followed by production of interferon (IFN)-γ by Th1 and natural killer cells. In normal infants the shift from Th2 to Th1 might be stimulated by exposure to a variety of antigens, including microbes with cytosine and guanosine nucleotides (CpG repeats). The hygiene hypothesis suggests that atopy develops because subjects are not sufficiently exposed to allergens at a young age, perhaps as a result of early use of antimicrobials or lack of exposure to allergens (e.g., children exposed to one another at day care, or environmental factors [e.g., farms]).28 The Th2 response that characterizes inflammatory disease is associated with increased production of IL-4, IL-5, and IL-13. The IL-4 from Th2 supports B-cell IgE synthesis. The Th2 cells have a particular influence on eosinophils through IL-5, which regulates both differentiation and bone marrow release (mature and immature cells), and the chemokine eotaxin.28 Subsequent accumulation of eosinophils in atopic tissue is influenced by continued recruitment by eotaxins, the presence of selective adhesion molecules, and delayed apotosis.28
The role of the bone marrow in the pathophysiology of atopy increasingly is emerging as potential target, leading to a systemic rather than local approach to therapy.34 Signaling appears to occur between the lung and bone marrow, with IL-5 and/or eotaxin being the commonality.35 Increased IL-3 may also play a role in eosinophil stimulation, whereas IL-18, IL-12, and INF-γ are inhibitory toward eosinophils.28,36 In animal models of asthma, including the canine Ascaris model, allergen exposure is associated with an increase in bone marrow eosinophils and trafficking from the bone marrow to the lungs.37 Interestingly, cysteinyl leukotrienes may influence eosinophil differentiation (in the presence of IL-3 or IL-5); leukotriene-receptor antagonists are able (in vitro) to suppress eosinophil differentiation.37,38
Eosinophils contribute to mucosal damage through release of basic amines, cysteinyl leukotrienes, and PAF.28 Inhibitory muscarinic (M2) receptors are damaged, which facilitates persistent bronchoconstriction. Mediators are released following binding of allergens with the α chain of high-affinity IgE receptor (Fc€ RIα) on the cell. Regulation of IgE, which is found both on and in eosinophils, offers another potential target of therapy. IL-4 and IL-13 are the most important inducers of IgE production.28
As permeability increases in response to inflammation, histamine and other inflammatory mediators are better able to reach and stimulate inflammatory cells located in the submucosa. The continued release of mediators is associated with stimulation of afferent nerve endings in the mucosa and reflex cholinergic bronchoconstriction. Mediators also increase microvascular permeability, induce chemotaxis, and stimulate mucous secretion. The release of cytotoxic proteins and toxic oxygen radicals further damages the respiratory epithelium, and the bronchial tree becomes hypersensitive. Mediators can also inhibit mucociliary function.25 Mucus production increases as submucosal glands and goblet cells increase. The consistency of the mucus changes to become more viscous. Bronchial smooth muscle often hypertrophies and undergoes spasms.39 Airway obstruction in chronic disease reflects bronchoconstriction, bronchial wall edema, and accumulation of mucus and cells. As the disease progresses, airways eventually become plugged and ultimately collapse. Chronic inflammation leads to fibrosis, which contributes to the collapse, and air trapped within the alveoli can result in emphysema. Asthma is a disease of central and large as well as small (<2 mm) airways.40 Regarding the type of asthma previously considered “silent,” studies in humans have revealed the importance of a therapeutic focus on airways beyond the eighth or ninth generation of the bronchial tree. In humans asthma and allergic rhinitis commonly, but not always, co-exist,40 and allergens associated with rhinitis may be too large to penetrate the lower airways.41
Inflammatory Mediators in the Respiratory Tract
Histamine
Histamine is a vasoactive amine stored in basophils and mast cells. Airway mast cells are located primarily beneath the epithelial basement membrane in dogs.24 Histamine produces a variety of effects (see Table 20-1) by interacting with specific receptors on target cells.12,42 At least four histamine receptors have been identified,5,29,43,44 two of which have been found in the trachea of the cat.5,6 Interaction with the H1 receptor causes an increase in intracellular calcium, and ultimately in cGMP (see Figure 20-3).29 Histamine also stimulates cholinergic receptors in the airway.24,29 Histamine causes constriction in both central and peripheral airways in dogs and cats.12,24 The effects of histamine so closely mimic the pathophysiology of early asthma that, for many years, histamine was considered the major cause of the syndrome.29 Lack of clinical response to H1-receptor antagonists, however, led to the realization that other factors are more important. In contrast to H1 receptors, stimulation of H2 receptors causes an increase in cAMP and bronchodilation.29 Thus antihistamine drugs that block H2 receptors may be contraindicated in asthma. Some studies have suggested that a defect in H2 receptors may contribute to airway hyperreactivity.29 Finally, recent evidence suggests that histamine regulates both Th1 and Th2 cells. Further, cells perpetuating the allergic response may be influenced by histamine through H4 receptors located on eosinophils, basophils, mast cells, and dendritic cells.43
Histamine contributes to bronchial occlusion by mechanisms other than bronchoconstriction. Mucous secretion is mediated via H2 receptors and by secretion of ions and water via H1 receptors.29 Microvascular leakage resulting from contraction of endothelial cells also follows H1-receptor stimulation.29 Histamine is chemotactic to inflammatory cells, particularly eosinophils and neutrophils. Interestingly, histamine stimulates T-lymphocyte suppressor cells by way of H2 receptors,5 a function that also may be depressed in human patients with asthma.29 Histamine also has a negative feedback effect on further histamine release mediated by IgE.29 Both of these latter effects are mediated by H2 receptors and would be inhibited by H2-receptor antagonists.5
Serotonin
Serotonin (5-hydroxytryptamine [5-HT]) is released during mast cell degranulation.29 Although serotonin does not appear to be an important mediator of human or canine bronchial asthma, both the central and peripheral airways in cats are very sensitive to its bronchoconstrictive effects after aerosolization or intravenous administration.12 Constriction may reflect interaction with serotonin receptors or enhanced release of acetylcholine. Serotonin may also cause profound vasoconstriction of the pulmonary vasculature and microvascular leakage.29
Prostaglandins and Leukotrienes
Prostaglandins (PGs) and leukotrienes (LTs) are eicosanoids that are formed when phospholipase A2 is activated in the cell membrane in response to a variety of stimuli (see Figure 20-3). Arachidonic acid (AA) is subsequently released from phospholipids and enters the cell. In the cell it is converted by cyclooxygenases to inflammatory, but unstable, cyclic endoperoxides. The actions of various synthetases and isomerases on the endoperoxides result in the final PG products, including PGE2, PGF2α, PGD2, prostacyclin (or PGI2), and thromboxane (TXA2). The amount of each PG produced in the lung varies with the cell type and species. The effects of the various PGs tend to balance one another. PGD2, PGF2α, and TXA2 cause bronchoconstriction, whereas PGE1 and, to a lesser extent PGI1, cause bronchodilation.1,29 Bronchoconstriction induced by PGD2 is about 30 times as potent as that induced by histamine.
Imbalances between PGs may be important in the pathogenesis of bronchial disease. Both PGD2 and TXA2 have been implicated in immediate bronchial airway hyperreactivity.29 Thromboxane A2 appears to be the predominant AA metabolite produced by feline lungs,45 although other PG mediators are also important.45
The role of LTs in atopy has been increasingly scrutinized in recent years. Lipoxygenases catalyze the conversion of AA to hydroperoxyeicosatetraenoic acid (HPETEs), which are further metabolized to several hydroxy acids (HETEs) and LTs (Chapter 29). Eosinophils have been known to be preferentially activated 5-lipoxygenase29; selective activation of lipoxygenase by antigenic challenge yields subsequent formation of the cysteinyl LTs (cystLTs: LTB4 C4, and D4), recognized as the components of slow reactive substance of anaphylaxis.29 However, both intermediate and end products of lipoxygenase are active in the respiratory tract (see Table 20-1).29 Among these mediators are some of the most potent inflammagens known. For example, bronchial smooth muscle contraction and microvascular permeability mediated by LTC4 and LTD4 is 100- to 1000-fold more potent than that induced by histamine. Both LTs are potent stimulators of mucous release in the dog but appear to be less potent in the cat.29 In humans airway allergen challenge increases LTs in urine and bronchoalveolar lavage (BAL) fluid with asthma and nasal secretions with allergic rhinitis.35 Increases in LTs in plasma, BAL fluid, or urine of challenged cats is controversial, although cats experimentally infected with dirofilariasis likewise exhibited increased plasma as well as BAL fluid cysLT concentrations (unpublished data). The cysteinyl LTs exert biologic effects through cysLT1 and cysLT2 receptors.35 Binding by LTs to eosinophilic cysL1 receptors (particularly LTD4) results in chemotaxis and prolonged survival, effects that are blocked by LT-receptor antagonists.35 Further evidence suggests that cysteinyl LTs are also involved in the initial systemic atopic response that is mediated from the bone marrow.35,46
Platelet-Activating Factor
PAF is also formed after activation of phospholipase A2 in cell membranes. It is a potent, dose-independent constrictor of human airways, and it is the most potent agent thus far discovered in causing airway microvascular leakage.22,23 PAF is also a potent chemotactant for platelets and eosinophils, both of which are a rich source of PAF. The effects of PAF may be mediated through LTs. PAF has been implicated as the cause of the sustained bronchial hyperresponsiveness that characterizes asthmatics.23 The role of PAF in feline and canine respiratory diseases has not been addressed. Eosinophils are, however, a major cell type associated with feline bronchial disease and some canine diseases, and it is likely that PAF is an important inflammatory mediator.
Reactive Oxygen Species
Reactive oxygen species (ROS) are constantly formed in the lung as part of pulmonary defense toward microorganisms and neoplasm.21 Free oxygen radicals, hydrogen peroxide and hypochlorous acid are among the species formed. Their formation is mediated, in part, by other inflammatory mediators such as TNF, IL-1, IL-6, and IL-8, and AA derivatives (PGs, LTs, and PAF). Hyperoxia will enhance generation of ROS.21 Normal cells are generally protected from ROS by a tightly controlled redox balance and formation of endogenous antioxidants; formation is enhanced by exposure to antioxidants. Those located primarily intraceullularly include catalase, superoxide dismutase, and glutathione redox compounds (glutathione, glutathione peroxidase, and glutathione reductase). Primarily extracellular antioxidants include fat-soluble compounds (vitamin E), water-soluble compounds (vitamin C, cysteine, reduced glutathione, taurine), and high-molecular-weight antioxidants (mucus and albumin).21
Neuropeptides
Several chemicals stimulate release of inflammatory neuropeptides from sensory neurons enervating smooth muscles. Cells stimulating neuropeptide release include macrophages, T-cells, eosinophils, and mast cells; example neuropeptides include substance P and, to a lesser degree, calcitonin gene-related peptide and neurokinin A.47 As with other inflammatory mediators, neuropeptides are associated with vasodilation, increased permeability and mucus production, as well as histamine release.
Cytokines
The role of cytokines in the coordination and persistence of chronic airway inflammation is integral and, not surprisingly, very complex. The integrated nature of their actions renders them simultaneously ideal as targets of drug therapy but potentially a source of adverse events should therapy be successful. Four cytokine classes have been proposed: lymphokines, chemokines, and proinflammatory and antiinflammatory cytokines (see Table 20-1). The role of lymphokines was largely addressed with the discussion of Th cell role in atopy. Initiation of Th2 cells is not clear, but appears to involve presentation of restricted antigens in the presence of IL-4 and IL-10. Perhaps the most notable lymphokine of asmtha is IL-5.48 Recruitment of eosinophils into the lungs—the hallmark of asthma—is IL-5 dependent. However, IL-5 also is integral to the differentiation, proliferation, and maturation of eosinophil progenitor cells in the bone marrow before recruitment into the lung.
Chemokines are cytokines that attract inflammatory cells, including eosinophils, monocytes, and T-lymphocytes. Two groups have been described based on proximity of cysteine residues to one another: CXC or α chemokines (separated by an amino-acid) and CC or β-chemokines (no separation; see Table 20-1). Chemokines exert their effects through rhodopsin-like G protein–coupled receptors (referred to as CXC-R or CC-R). Because certain cells express selected receptors, pharmacologic therapy increasingly will be designed to be selectively antiinflammatory.31 Among the cells influenced by chemokines are eosinophils, which in turn release more chemokines. Among the most notable drugs targeting proinflammatory chemokines, and particularly their transcription, are glucocorticoids (see Chapter 30). Newer therapies take advantage of the presence of antiinflammatory cytokines; indeed, glucocorticoids facilitate restoration of antiinflammatory cytokines such as IL-10.
Communication Molecules: Adhesion Molecules and Integrins
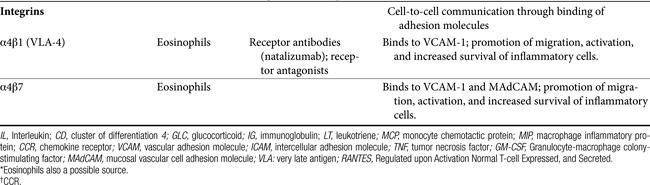
Communication between cells is facilitated by adhesion molecules that interact, by way of ligand binding, with receptors on the surface of inflammatory cells known as integrins. Integrins are present on many cells, with receptor specificity occurring for selective cells. Integrins are absent on Th1 cells but are particularly prevalent on Th2 cells, mast cells, and eosinophils. Interaction between adhesion molecules and integrins promotes migration, activation, and increased survival of inflammatory cells. Drugs that target eosinophilic integrins (α4β1 and 7) offer a possible mechanism whereby selective control of inflammation may limit side effects.49
Drugs Used to Modulate the Respiratory Tract
Bronchodilators and Anti-Inflammatory Drugs
Because of a shared intracellular mechanism of action, most drugs that induce bronchodilation also reduce inflammation. Bronchodilators reverse airway smooth muscle contraction by increasing cAMP, decreasing cGMP, or decreasing calcium ion concentration (see Figure 20-1). In addition, these drugs also decrease mucosal edema and are antiinflammatory because they tend to prevent mediator release from inflammatory cells (see Figure 20-3). Rapidly acting bronchodilators include β-receptor agonists, methylxanthines, and cholinergic antagonists.
β-Receptor Agonists
β-receptor agonists (Figure 20-4) are the most effective bronchodilators because they act as functional antagonists of airway constriction, regardless of the stimulus.22,50,51 Few β-agonists generally have been sufficiently studied in animals to describe pharmacokinetics or pharmacodynamics.
Large numbers of β2-receptors are located on several cell types in the lung, including smooth muscle and inflammatory cells.3 The receptor is linked to a stimulatory guanine nucleotide-binding protein (G protein). The interaction between a β-agonist and receptor causes a conformational change in the receptor and subsequent activation of adenylyl cyclase on the inner cell membrane (see Figure 20-1).52 Adenylate cyclase converts adenosine triphosphate to cAMP, a second messenger for activation of specific protein kinases that ultimately activate enzymes responsible for airway smooth muscle relaxation. β-receptor agonists are most effective in states of bronchoconstriction. Additional effects of β-adrenergic receptors include increased mucociliary clearance, which reflects a decrease in fluid viscosity (presumed to reflect movement of chloride and water into the lumen) and an increase in ciliary beat frequency. Additionally, they inhibit cholinergic neurotransmission, enhance vascular integrity, and inhibit mediator release from mast cells, basophils, and other cells (see Figure 20-3).22,23,51,52 Eosinophil, but not lymphocyte, numbers appear to decrease, but long-term inflammation does not appear to be affected,52 probably because inflammatory cell β receptors are rapidly desensitized (discussed later). As such, long-term β-adrenergic use should be accompanied by antiinflammatory therapy.41
As with many membrane-associated receptors, high doses or repeated exposure to β-adrenergic agonists results in desensitization.51 The mechanism depends on the duration of therapy. Initial drug–receptor interaction causes phosphorylation, which interferes with G proteins. Longer exposure causes receptors to internalize such that they are not accessible by the drug; continued exposure causes downregulation of receptor mRNA such that the number of receptors is actually reduced.52 Reduction develops over several weeks before stabilizing. Desensitization may contribute to acute exacerbations of bronchoconstriction associated with long-acting β-adrenergics. Bronchodilation will be less in the desensitized airway necessitating increased treatment frequency. However, frequent use may mask clinical signs associated with uncontrolled inflammation and care must be taken that long-acting β-adrenergics are not used instead of antiinflammatory therapy. Despite these shortcomings, β-adrenergics are the most the most effective bronchodilators.
The β-adrenergic agonists (see Figure 20-4) can be given by a variety of routes. In humans inhalation is preferred because it is equally effective to parenteral administration and is safer. However, drug delivery to the peripheral airways must be addressed as much as possible.
Nonselective β-Agonists
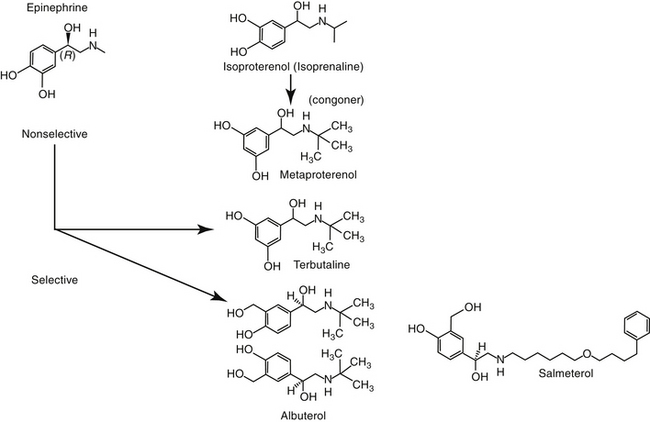
The nonselective β-agonists (i.e., capable of both β1 and β2 stimulation) such as epinephrine, ephedrine, and isoproterenol are used for acute and chronic therapy of respiratory diseases. Epinephrine and isoproterenol can be administered parenterally to achieve rapid effects, and drugs that can be given orally for chronic therapy include isoproterenol and ephedrine.6 Both epinephrine and ephedrine cause α-adrenergic activity, which may cause vasoconstriction and systemic hypertension and may contribute to airway constriction.4 Nonselective β-agonists may cause adverse cardiac effects as a result of β1-receptor stimulation. Aerosolization reduces the adverse effects of nonselective β-adrenergic agonists by increasing β2 specificity, because only these β-receptors appear to line the airways.
β2-Selective Agonists
At appropriate doses, β-selective agonists are not generally associated with the undesirable effects of β1-adrenergic stimulation. The more commonly used β-agonists are categorized as to their duration of action: intermediate (3 to 6 hours) and long-acting (>12 hours). The two long-acting bronchodilators, salmeterol and formoterol, both have extended side chains, are highly lipophilic, and are characterized by high affinity for the β-2 receptors. However, the long duration of salmeterol reflects binding to a specific site within the receptor, whereas that for formoterol reflects gradual release from the cell membrane lipid.52 The long-acting nature of bitolterol reflects its pulmonary metabolism to an active metabolite. However, it has a rapid onset of action and generally is used for acute therapy.
Short-acting drugs that have been used in animals include albuterol, metaprotereno (a derivative of isoproterenol), and its analog terbutaline.41 Metaproterenol is less β-2 selective than other agents and more apt to cause cardiac stimulation.52 Therefore repetitive use should be pursued only cautiously. Albuterol, metaproterenol and terbutaline are available as oral preparations; each has been used apparently safely in dogs. Rapid first-pass metabolism reduces systemic bioavailability after oral administration; as such, oral doses are greater than parenteral doses. These drugs, but particularly metaproterenol, can cause β1 side effects at high doses. Clenbuterol, also a β-2 selective agonist, is approved for use in horses. Although manufactured as the racemic mixture, pharmacologic effects are largely limited to the L-isomer.53 Tachycardia occurs in dogs at 0.4 mg/kg, which is only half of the therapeutic dose recommended in horses. Further, cardiac necrosis occurs at 2.5 mg/kg, which represents only a threefold increase over the equine therapeutic dose. Consequently, use of clenbuterol in dogs may not be prudent. Albuterol and isoetharine are examples of β2-selective agonists that have been administered by aerosolization to small animals.30 Salbutamol is frequently used as a rescue bronchodilator in humans.54
Inhalant Devices
With the advent of metered doses inhalers in the 1960s, beta-adrenergics became a common therapy for treatment of human asthma. Short-acting β2 agonists administered by aerosol (Figure 20-5) include albuterol and its R enantiomer levalbuterol, metaproteronol, pirbuterol, bitolterol, and terbutaline (see Figure 20-4). Longer-acting (in humans) beta-2 adrenergic drugs tend to be more lipophilic and thus remain in the presence of a receptor for a longer time. Examples include salmeterol and formoterol (see Figure 20-4).40,41,55 The drugs differ in their effect and use. Short-acting products are associated with rapid symptomatic relief in human asthmatics when used at appropriate doses. However, use at high doses has been associated with an increase in mortality in humans, leading to their recommended use on an “only as needed” basis.40,55 On the other hand, improvement in pulmonary function in humans was sustained with prolonged used of long-acting beta-adrenergics,55 and thus such use was not associated with a decrease in symptomatic relief afforded by short-acting drugs. The duration of onset of long-acting beta-adrenergics may be 1 or more hours. Although minimally effective by themselves for control of inflammation, long-acting beta-adrenergics appear to enhance responsiveness to glucocorticoids. Rebound hyperresponsiveness does not appear to occur with rapid discontinuation of the long-acting drugs. The development of tolerance (desensitization) has been discussed and is probably more likely with long-acting drugs.41 Because beta-adrenergics do not provide as much antiinflammatory control, their efficacy may be reduced in the presence of inflammation and combination therapy with an antiinflammatory drug (e.g., inhaled or systemic glucocorticoids), or use of theophylline may be indicated.40,55

Methylxanthine Derivatives
Pharmacologic Effects
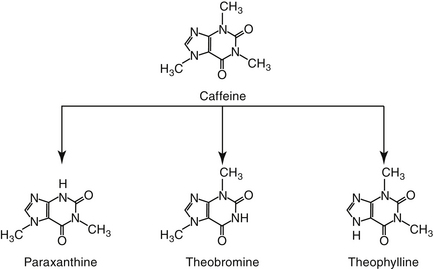
Methylxanthines include caffeine, a potent respiratory stimulant, and theobromine, a recognized canine toxicant from chocolate and theophylline (Figure 20-6). Theophylline has been the cornerstone of long-term bronchodilatory therapy in animals, particularly dogs. Its mode of action has been attributed to nonspecific inhibition of phosphodiesterases (PDEs) and increased concentrations of cAMP and cGMP (see Figure 20-1).41,56 This mechanism has been controversial, however, because theophylline does not inhibit PDE at therapeutic concentrations. However, this may reflect the existence of PDE as various isoenzymes (at least 11 members to this superfamily, each located in different sites within the cell, some of which are inaccessible to drugs).22,23,41 Inhibition of PDE4 and PDE5 results in bronchodilation whereas inhibition of PDE4 contributes to its antiinflammatory effects.41 The nonselective action of theophylline results in bronchodilation and inhibition of inflammation. Theophylline also is a competitive antagonist at adenosine receptors. The inhibitory neurotransmitter adenosine, which induces bronchoconstriction and during hypoxia and inflammation. Another mechanism by which theophylline induces bronchodilation, however, may be through interference of calcium mobilization.22,23
Compared to beta-2 agonists, theophylline is considered a weaker relaxant of airway smooth muscle.40 As with β-agonists, theophylline is equally effective in large and small airways. Theophylline has other effects in the respiratory system that are important to its clinical efficacy.22,23,56 In addition to its bronchodilatory effects, it inhibits mast cell degranulation and thus mediator release (see Figure 20-3)57; increases mucociliary clearance; and prevents microvascular leakage.58 Its antinflammatory effects appear to include modulation of cytokines, particularly of macrophage origin.59 Finally, antiinflammatory effects of theophylline may also reflect nuclear activation of histone deacetylase.41 Decreased TNF-α and IFN-γ and increased IL-10 (antiinflammatory) have been reported. Pentoxifylline has similar antiinflammatory effects; its IV administration is addressed in Chapter 29. Increased mucociliary clearance has been reported in dogs.60 In propofol-anesthetized cats (n = 6), no treatment effect could be detected in the mucociliary transport rate after oral administration of 25 mg/kg of a slow release (Slo-bid) product. The ability to detect a significant difference was not addressed. Further, the mucociliary the rate in untreated animals (22.2 ± 2.8 mm/min) was perceived to be maximal, leaving no room for theophylline-induced increase. The impact of propofol is not clear, and theophyllline concentrations were not measured to confirm adequate concentrations. Thus the lack of efficacy may not reflect theophylline as much as failed delivery or altered response.
In addition to its antiinflammatory effects, a major advantage of theophylline, compared with other bronchodilators, may be its increased strength of respiratory muscles and thus a decrease in the work associated with breathing.56,62,63 This may be important to animals with chronic bronchopulmonary disease.
Disposition
Theophylline is one of the few drugs active in the respiratory tract whose disposition has been studied in animals. Because theophylline is not water soluble, it can be given only orally. Salt preparations of theophylline are available for either oral or parenteral administration. Dosing of the various salt preparations must be based on the amount of active theophylline (Table 20-2). Aminophylline, an ethylenediamine salt, is 80% theophylline, whereas oxtriphylline is 65% theophylline, and glycinate and salicylate salts are only 50% theophylline. Regular aminophylline is well absorbed (bioavailability of at least 90%) after oral administration in both dogs and cats.64,65 In dogs peak plasma drug concentrations for the theophylline base (approximately 8 μg/mL after a dose of 9.4 mg/kg) occur 1.5 hours after oral administration.64 The volume of distribution (L/kg) and clearance (mL/min/kg) in dogs are, respectively, 0.59 ± 0.045 and 0.78 ± 0.13. The extrapolated peak concentration after intravenous administration of 11 mg/kg aminophylline (8.6 mg/kg theophylline equivalent) was 22.94 ± 5.8 μg/mL.66 In cats extrapolated concentrations after 10 mg/kg intravenous aminophylline approximated 10 μg/mL; volume of distribution (L/kg) and clearance (mL/min/kg) were 0.87 ± 0.07 and 0.87 ± 0.16, respectively.67 Interestingly, peak concentrations after intravenous or oral (sustained-release) products are higher in cats when dosed in the evening compared with the morning.68
More than 30 slow-release preparations exist for use in humans. Although several products have been studied in dogs and cats,68,69 their disposition is markedly variable and cannot be predicted on the basis of product name or description. Interchangability of products cannot be assumed, and monitoring is strongly encouraged to establish potential efficacy. For dogs, the rate of oral absorption of slow-release products is apparently faster than in humans. The extent of absorption varies with the preparation. Bioavailability of slow-release preparations varies from 30% (anhydrous theophylline 24-hour capsules; Theo-24, Searle Laboratories) to 76% (anhydrous theophylline tablets; TheoDur).69 The least variation among animals occurs for oxtriphylline enteric-coated capsules (Choledyl-SA Tablets; Parke-Davis) and a 12-hour capsular anhydrous theophylline (Slo-Bid Gyrocaps), which are approximately 60% bioavailable. The minimum effective concenteration recommended for humans (10 μg/mL) may not be reached by all slow-release products. Plasma drug concentrations during a 12-hour dosing interval varies, by almost 120% for the oxtriphylline product but only 48% for the anhydrous tablet (Theo-Dur), suggesting it may be the best product for use in dogs.69 However, neither Theo-Dur nor Slo-Bid Gyrocaps are currently available for use in the United States. Although the mean residence time of the slow-release preparation was significantly longer by 1 to 2 hours than that of the regular preparation in dogs, the clinical significance of this difference is questionable.69 More recently, Bach and coworkers66 described the disposition of a generic (Inwood Laboratories) extended-release theophylline tablet (Theochron) or capsule (TheoCap) in dogs (15.5 mg/kg orally; n = 6). The oral bioavailability (%) of the tablet and capsules was 98 ± 15.4 and 83.6 ± 18.5%, respectively. The Cmax (μg/mL) was 17.4 ± 4.9 and 12.2 ± 2.8 for the tablet and capsule, respectively, whereas the elimination half-life was 10.9 ± 3.6 and 12.7 ± 2.7. Drug concentrations remained within the recommended therapeutic range for 12 hours. The authors concluded that 10 mg/kg every 12 hours would result in theophylline concentrations in the therapeutic range.
Cats also have been studied. Although it is not distributed to all body tissues, theophylline is characterized by a relatively large volume of distribution in dogs (0.7 to 0.8 L/kg) and a smaller volume in cats (0.41 L/kg).64,69,70 Unlike human beings, distribution of theophylline is not limited by binding to serum proteins in dogs; serum protein binding is less than 12%.71,72 Elimination of theophylline is not dose dependent in dose ranges of 3 to 15 mg/kg. Dye and coworkers68,70 studied two sustained-release theophylline products (TheoDur and Slo-Bid Gyrocaps, William H. Rorer, Inc) in the cat. Although both products were reasonably (>75%) bioavailable in the cat, neither product is currently available in the United States. A more recent study addressed the disposition of a generic (Inwood Laboratories) extended-release theophylline tablet (Theochron; 15 mg/kg) and capsule (TheoCap; 19 mg/kg) in cats (n = 6).67 The Cmax (μg/mL) of the tablet and capsule was 17.8 ± 3.4 and 15.8 ± 3.1, respectively. Although bioavailability of both products was 100% or greater, the elimination half-life for the tablet and capsule was 13.6 ± 3.9 hour and 18.3 ± 8.0, respectively, compared with 11.7 ± 1.8 hour for the intravenous preparation (aminophylline), suggesting that the drugs did not always act in an extended-release manner in cats. Nonetheless, drug concentrations remained within the recommended therapeutic range for 24 hours. These data suggested that once-daily administration in cats should be sufficient to maintain concentrations within the therapeutic range. (see Table 20-2). A longer dosing interval might be acceptable if evidence supports an antiinflammatory effect at even lower concentrations. Based on a chronopharmacokinetic study of these sustained-release products, dosing in the evening rather than in the morning appears to be associated with better bioavailability and less peak plasma theophylline concentration fluctuation.68 A disadvantage of the use of the slow-release products for small animals is the limited dose sizes available. The product cannot be divided for more accurate dosing without altering the kinetics of slow release.
Theophylline is metabolized by demethylation in the liver. Theobromine may be an active metabolite in some species. Different rates of metabolism result in variable clearance rates and drug elimination half-lives among animals, and doses consequently vary.72 For example, the elimination rate constant of theophylline is less in cats (0.089/hr)73 than dogs (0.12/hr), resulting in a longer half-life in the cat (7.8 hours) compared with the dog (5.7 hours),64,65 thus necessitating a smaller dose in cats.69 Theophylline concentrations can be affected—most commonly increased—by a number of drugs, including fluorinated quinolones,74 erythromycin and its congeners,75 and cimetidine.76
Adverse Reactions and Drug Interactions
Theophylline is associated with a wide range of adverse effects, including central nervous system excitation (manifested as restlessness, tremors, and seizures),72 gastrointestinal upset (nausea and vomiting), diuresis, and cardiac stimulation (e.g., tachycardia). In a retrospective study of theophylline toxicity in human patients, clinical signs included severe vomiting (89%), seizures (21%), and cardiac arrythmias (16%).77 Vomiting precluded treatment with oral charcoal but responded to metoclopramide. Therapy included mechanical ventilation, anticonvulsants or sedatives, and muscle relaxants. Because of the risk of toxicity, intravenous use should be limited to patients who have not responded to β-agonist therapy and are facing life-threatening disease. Compared with the salt preparations, theophylline is more irritating to the gastrointestinal tract than aminophylline.30,56 Rapid infusions or infusions of undiluted aminophylline can cause cardiac arrhythmias, hypotension, nausea, tremors, and acute respiratory failure.30
Theophylline may be involved in a number of drug interactions; hepatic metabolism and potentially serious adverse effects should lead to cautious use with other drugs. Inhibition of drug-metabolizing enzymes by fluorinated quinolones has been described in human medicine and for both enrofloxacin and marbofloxacin in animals.78 For enrofloxacin 5 mg/kg decreased theophyllline clearance by 43%79; a different study found a 26% decrease by marbofloxacin (5 mg/kg).78 This translates to an increase in Cmax by 31% for theophylline, sufficient to contribute to toxicity. Indeed, the author has measured peak theophylline concentrations above 70 μg/mL and an elimination half-life of 19 hours in a dog concomitantly given enrofloxacin at 5 mg/kg. Clinical signs of theophylline toxicity emerged within 24 hours of beginning enrofloxacin therapy. Theophylline–terbutaline interactions do not appear to be clinically relevant, as has been shown in humans,80 supporting their combined use for patients not responding to a single bronchodilator.
The application of therapeutic drug monitoring to guide therapy would assist in identifying the most appropriate dosing regimen, particularly when using slow-release preparations whose bioavailability might vary among patients, in patients receiving drug combinations that increase the risk of drug interactions, and in patients with hepatic dysfunction. Although a therapeutic range has not been established for small animals, the range recommended for humans (10 to 20 μg/mL) can be extrapolated until a more definitive range has been established. Dogs are apparently more tolerant of theophylline toxicity than are humans. In one study toxicity manifested as tachycardia, central nervous system stimulation (restlessness and excitement), and vomiting did not occur until plasma theophylline concentrations reached 37 to 60 μg/mL. Doses of 80 to 160 mg/kg of a sustained-release preparation were required to induce toxicity.81 In cats concentrations as high as 40 μg/mL do not induce adverse reactions, although salivation and vomiting are common after administration of more than 50 mg/kg, and seizures may occur at doses greater than 60 mg/kg.82
The side effects of theophylline are dose dependent and might be prevented to a large degree by appropriate dosing. Therapeutic drug monitoring should facilitate the design of proper dosing regimens to prevent toxicity.
Anticholinergic Drugs
Pharmacologic Effects
Anticholinergic drugs compete with acetylcholine at muscarinic receptor sites.83 In the respiratory tract, they reduce the sensitivity of irritant receptors and antagonize vagally mediated bronchoconstriction. The site of action of these drugs in the respiratory tract is controversial. In some studies bronchodilation is reported throughout the airways in asthmatic human patients and cats, whereas other investigators believe that the effects are confined to large airways.83 The route by which anticholinergic drugs are administered influences their bronchodilatory effects. Despite their effect on bronchial airways, the anticholinergic drugs have not proved clinically effective in the treatment of bronchial diseases in animals, and their use is limited to treatment of bronchoconstriction associated with organophosphate toxicity or in animals in status asthmaticus unresponsive to bronchodilator therapy. The lack of clinical efficacy of anticholinergics may reflect nonselective drug–receptor interaction.22,23 Thus far, three types of muscarinic receptors have been identified in airways. M3 receptors release acetylcholine, whereas M2 receptors block its release. Nonselective blockade of muscarinic receptors by atropine and ipratropium may actually potentiate acetylcholine release by antagonizing the effects of M2-receptor stimulation. Drugs specific for M3 receptors may ultimately lead to successful treatment of bronchial disease with anticholinergics drugs.22,23 As with beta-adrenergic drugs, use of inhaled anticholinergic drugs reduces systemic adverse effects.
Atropine
Aerosolized atropine, a prototype anticholinergic drug, affects predominantly the central airways, whereas both central and peripheral airways are affected if the drug is administered intravenously.22,23 Because atropine is highly specific for all muscarinic receptors, it causes a number of systemic side effects, including tachycardia, meiosis, and altered gastrointestinal and urinary tract function.64 In the respiratory tract, atropine reduces ciliary beat frequency, mucous secretion, and electrolyte and water flux into the trachea. The net effect is decreased mucociliary clearance, which is undesirable in patients with chronic lung disease.64 Aerosolization of atropine does not reduce the incidence of respiratory adverse reactions. Atropine is well absorbed (in humans) after oral administration. In humans atropine has proved most useful for treatment of chronic bronchitis and emphysema, diseases that are characterized by increased intrinsic vagal tone.83 Its adverse effects on respiratory secretions and ciliary activity, however, apparently negate its benefits to bronchial tone during long-term administration in animals. The primary indication for atropine in small animals is facilitation of bronchodilation in acutely dyspneic animals. It is the treatment of choice for life-threatening respiratory distress induced by anticholinesterases. A combination of atropine with either β-adrenergic agonists or glucocorticoids will cause better bronchodilation than either drug alone.83
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


