CHAPTER 16 Disorders of the Liver
NORMAL LIVER
Anatomy
The liver is the largest organ in the body, constituting approximately 1% of the body weight in the adult horse.1 The location of the liver between the gastrointestinal tract and the heart is functionally suited for its metabolic, secretory, excretory, and storage properties. In the normal horse, the liver lies mostly to the right of the median, is completely contained within the rib cage, and does not contact the ventral abdominal floor. The most cranial portion of the liver is located in the ventral third of the sixth to seventh intercostal spaces and extends caudad to the right kidney (fifteenth rib). In disease processes resulting in hepatomegaly, and in the normal equine neonate, the liver may extend beyond the caudal border of the last rib. Right liver lobe atrophy has been described as an uncommon normal anatomic variation in adult horses. However, in 1994 it was hypothesized that right hepatic lobe atrophy in horses is a pathologic condition resulting from long-term compression of the right lobe of the liver by abnormal distension of the right dorsal colon and base of the cecum.2
The equine liver consists of two surfaces, diaphragmatic and visceral, and is divided by fissures into four lobes: right, left, quadrate, and caudate. The visceral surface of the liver in situ is malleable and contains impressions of the organs with which it is in contact. The visceral surface also contains the hilum, or porta (door), of the liver, through which blood vessels, lymphatics, and nerves enter, and the hepatic duct exits. In the horse, six ligaments secure the liver in the abdominal cavity.1 The coronary ligament has two laminae, right and left, which attach the diaphragmatic surface of the liver to the caudal vena cava and the abdominal esophagus. The two laminae of the coronary ligament unite ventrally to form the falciform ligament. The falciform ligament, a remnant of the fetal ventral mesentery that extends from the diaphragm to the umbilicus, attaches the quadrate and left lobes to the sternal diaphragm and ventral abdominal floor. The round ligament, the remnant of the fetal umbilical vein, is contained within the free border of the falciform ligament. The right and left triangular ligaments attach the dorsal right lobe to the right costal diaphragm and the dorsal left lobe to the tendinous center of the diaphragm. The hepatorenal ligament connects the caudate process of the quadrate lobe to the right kidney and the base of the cecum.
Histology
At the hilum of the liver, a tree of connective tissue consisting of collagen and fibroblasts enters the hepatic parenchyma. The parenchymal cells, or hepatocytes, compose approximately 50% to 60% of the mass of the liver and are epithelial cells.3,4 The hepatocytes are arranged in rows, or cords, at least two cells thick that anastomose to form blood passageways called sinusoids (Figure 16-1). Hepatic sinusoids are larger than capillaries and are lined with endothelial cells and Kupffer’s cells. Kupffer’s cells are tissue-fixed macrophages and are estimated to make up 20% of the mass of the liver.4 The endothelial cells make up approximately 20% of the mass of the liver.4 A cleft, called the space of Disse, lies between the hepatocytes and the cells lining the sinusoids. The space of Disse contains fluid similar to the composition of blood but does not contain erythrocytes.
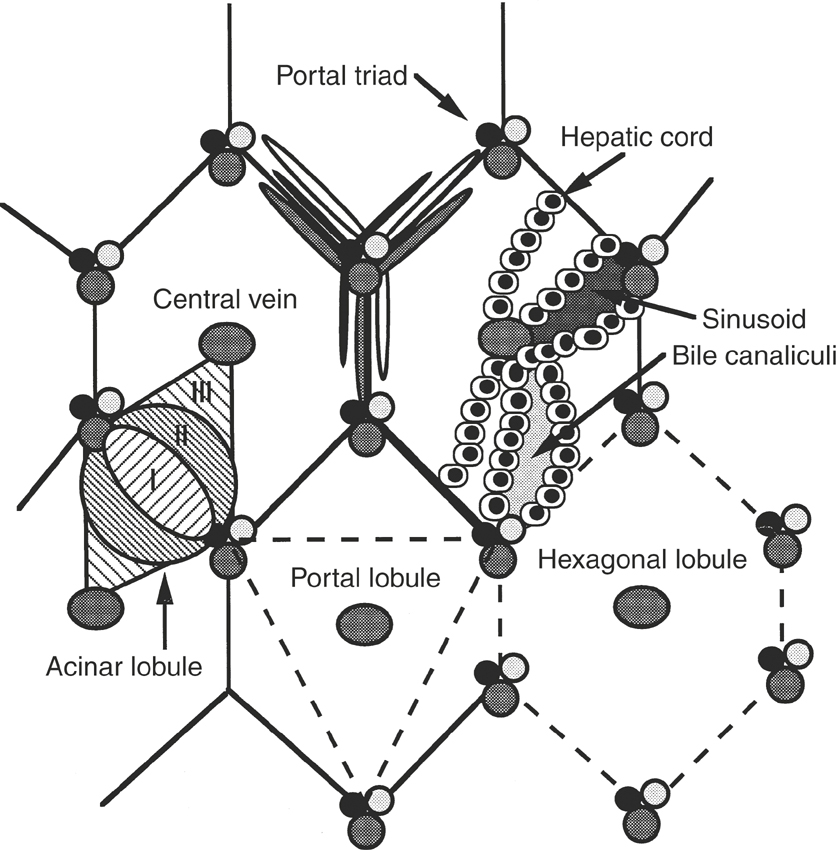
The space between contiguous hepatocytes in a cord forms a bile canaliculus through which bile excreted by the hepatocytes drains into bile ductules and ducts. The bile canaliculi thus are formed solely by the cell membranes of the hepatocytes. The bile ductules and ducts are lined with cuboidal and columnar epithelial cells, respectively, that make up approximately 7% of the mass of the liver.4 The bile ducts run in the connective tissue tree, adjacent to branches of the portal vein and hepatic artery, to form a distinct portal tract, radicle, canal, or triad (see Figure 16-1). The bile ducts converge at the hilum to form the hepatic duct, which drains into the duodenum just distal to the pylorus. Because the horse does not have a gallbladder or a sphincter at the entry site of the hepatic duct into the intestine, the bile is unconcentrated and flows continuously in a direction opposite that of the blood flow in the portal vein and hepatic artery.1
The liver can be divided anatomically or functionally into lobules to facilitate histopathologic description of lesions3 (see Figure 16-1). The classic hepatic lobule is delineated by abundant interlobular connective tissue that in cross-section roughly appears hexagonal. Three to eight portal tracts define the corners of the hexagon with a central vein in the center of the lobule. In contrast, the portal lobule is a functional unit describing the exocrine duties of the liver. Central veins define the three corners of the portal lobule, with a portal tract situated in the center. The acinus lobules describe the vascular supply to the hepatic parenchyma, divided according to the tissue oxygen content. Zone I of the acinus lobule is located immediately adjacent to branching hepatic arteries and portal veins, is the most metabolically active zone, and receives the best oxygen supply. Zone III is located adjacent to central veins, has high mixed-function oxidase activity, is least favorably situated with respect to oxygen content, and thus is most susceptible to toxic and hypoxic damage. Zone II is situated between zones I and III.
Physiology
The liver is the main organ involved in the regulation of nutrient distribution.5 The majority of nutrients absorbed from the gastrointestinal tract pass directly to the liver via the portal circulation. The incoming nutrients are metabolized for energy, transformed to other nutrient classes, packaged and exported to peripheral tissues, or stored by the liver. The liver is capable of adjusting to the carbohydrate, protein, and lipid load from the gastrointestinal tract, as well as maintaining consistent blood levels of nutrients between feedings and in response to special needs. In addition to its role in nutrient metabolism and homeostasis, the liver is involved in excretion (bile), detoxification and metabolism of endogenous and exogenous substances, and hematopoiesis.4
Protein Metabolism
Amino acids, which are transported to the liver via the portal or hepatic blood, may be used in the biosynthesis of intrinsic hepatocellular proteins, plasma proteins, porphyrins, polyamines, purines, and pyrimidines.5 The liver synthesizes 90% of the plasma proteins, including albumin, factors involved in coagulation and fibrinolysis (fibrinogen and factors II, V, VII-XIII; antithrombin III; protein C; plasminogen; plasminogen activator inhibitor; α2-antiplasmin; α2-macroglobulin; and α1-antitrypsin), transport proteins (haptoglobin, transferrin, ceruloplasmin, hormone transport proteins), and acute phase reactant proteins (α- and β-globulins).4 The liver is the only site of synthesis of albumin and fibrinogen.
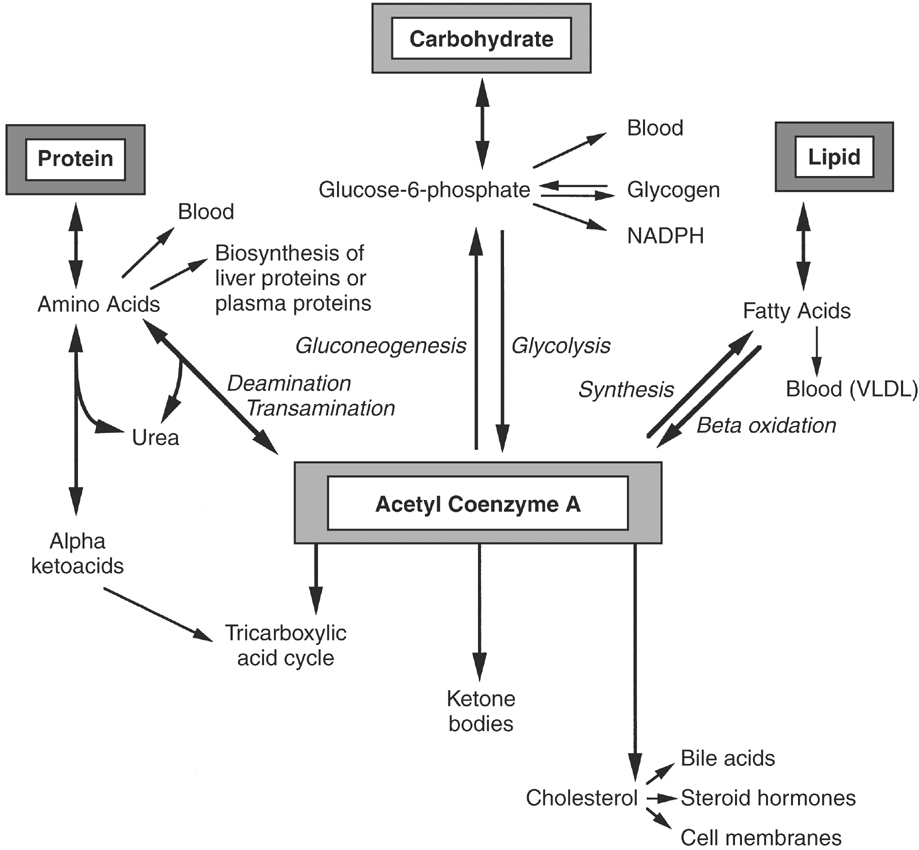
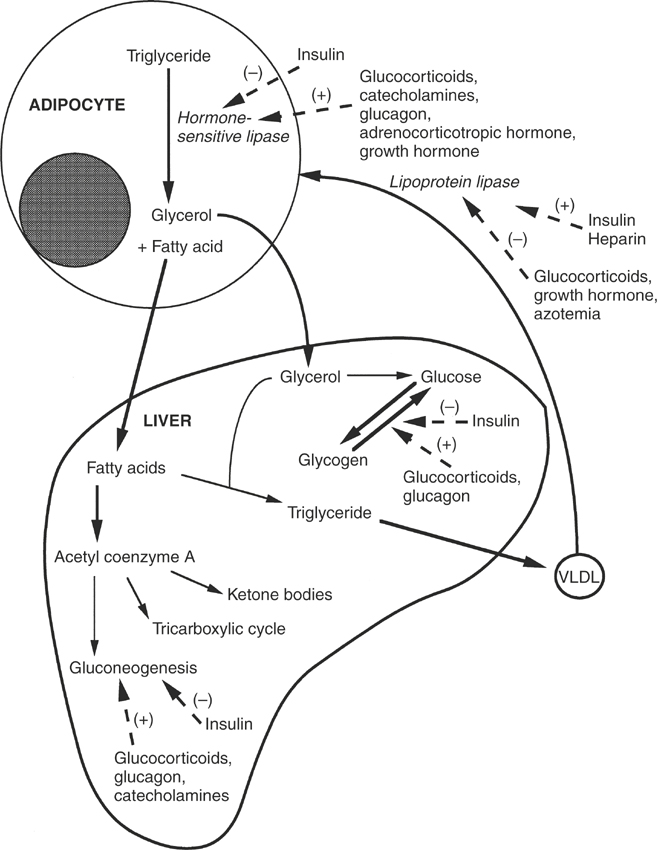
The liver is also capable of transamination, or the reversible transfer of an amino group on one amino acid to an α-keto acid, thus forming a new amino acid and a new keto acid. If the liver receives an excess of amino acids or if carbohydrates are unavailable as an energy source, then the liver deaminates the amino acids and converts them to pyruvate, acetoacetate, and intermediates of the tricarboxylic acid cycle5 (Figure 16-2). These intermediates may be oxidized for energy or used as precursors in gluconeogenesis, the synthesis of glucose from noncarbohydrate precursors. Endogenous and exogenous glucocorticoids, glucagon, and thyroid hormone act directly on the liver to increase gluconeogenesis6 (Figure 16-3). Simultaneously, glucocorticoids indirectly influence liver gluconeogenesis by promoting peripheral protein catabolism, thus increasing the availability of amino acids. Insulin inhibits gluconeogenesis in the liver.6
In addition to protein synthesis and gluconeogenesis, the liver plays an important role in eliminating the major toxic by-product of amino acid catabolism, ammonia.5,7 Tissues and intestinal microflora generate ammonia, which is subsequently released into the circulation. One method by which the liver, as well as certain peripheral tissues, eliminates ammonia is by synthesizing nonessential amino acids from α-keto acids and ammonia in a reversal of deamination. A fundamental reaction in the synthesis of nonessential amino acids is the formation of glutamate from α-ketoglutarate and ammonia (Figure 16-4). Subsequently, glutamate is used in transamination reactions to form other amino acids. Glutamate also participates in the conversion of cytotoxic free ammonia into a nontoxic transport form, glutamine. Glutamine may be delivered to the kidney, converted back to free ammonia and excreted, or delivered to the liver for urea synthesis.
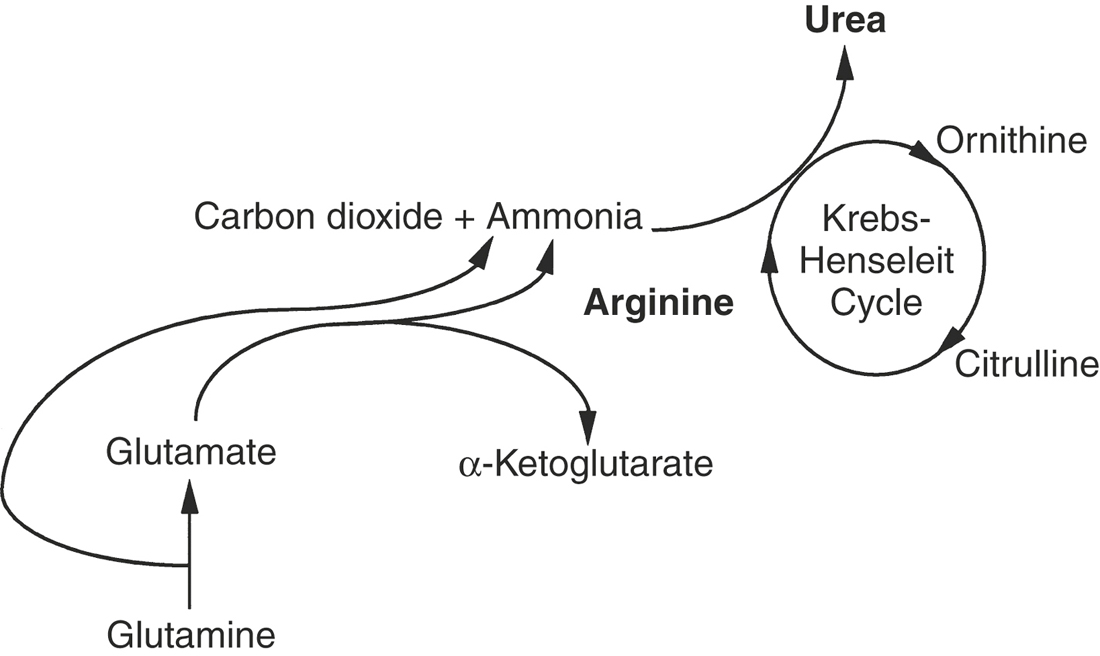
The liver has sole responsibility for converting free ammonia or glutamine into urea, the principal form of amino group nitrogen excretion by mammals.4 Urea is formed by the irreversible condensation of two ammonia molecules with carbon dioxide (see Figure 16-4). The reaction takes place in the hepatocyte mitochondria via the Krebs-Henseleit cycle.5 The newly formed urea is released from the hepatocyte, secreted into the sinusoidal blood, and transported to the kidney as blood urea nitrogen (BUN) for excretion.
CARBOHYDRATE METABOLISM
The liver is responsible for the synthesis, storage, and release of glucose.5 Monosaccharides absorbed from the gastrointestinal tract are delivered via portal blood to the liver. In the hepatocyte the majority of glucose is phosphorylated to glucose-6-phosphate by the enzyme hexokinase (see Figure 16-2). The remaining glucose is released into the systemic circulation. Other monosaccharides (fructose, galactose) are phosphorylated and converted in the liver to glucose-6-phosphate. The majority of glucose-6-phosphate is converted to glycogen for storage. A small amount of glucose-6-phosphate is oxidized to form adenosine triphosphate, although the major source of adenosine triphosphate in the liver is amino acid and fatty acid oxidation. Approximately half of the liver glucose enters the phosphogluconate pathway for generation of nicotinamide adenine dinucleotide phosphate, which is required as a reducing agent in the biosynthesis of fatty acids and cholesterol. Glucocorticoids, catecholamines, glucagon, and thyroid hormone increase gluconeogenesis and glycogenolysis in the liver, while insulin inhibits gluconeogenesis6 (see Figure 16-3).
LIPID METABOLISM
Short-chain fatty acids (fewer than 10 carbon atoms) can be absorbed directly from the gastrointestinal tract, bound to albumin, and delivered to the liver via the portal circulation.5 However, the majority of short-chain fatty acids are incorporated into phospholipid or triglyceride by the intestinal epithelium and transported to the liver via the portal blood. The remaining fatty acids absorbed from the gastrointestinal tract are transported as triglyceride in chylomicrons. After formation in the intestinal epithelial cells and absorption into lymphatics, chylomicrons enter the systemic circulation via the thoracic duct and subsequently are delivered to the liver. The liver also may take up albumin-bound fatty acids released from adipose tissue.
The fate of fatty acids in the liver depends on the state of energy demand, the rate of fatty acid delivery, and hormonal influences. The primary role of the liver in lipid metabolism is to esterify free fatty acids into triglycerides for export to other tissues5 (see Figure 16-3). The triglycerides are packaged with protein, carbohydrate, and cholesterol in the endoplasmic reticulum of the hepatocyte into very-low-density lipoproteins (VLDLs), which primarily contain triglyceride, and high-density lipoproteins (HDLs), which primarily contain protein and phospholipid.8 The VLDLs and HDLs are released into the hepatic sinusoids. Once the VLDLs are in the systemic circulation, the adipose tissue takes them up or endothelial cell lipases alter their composition by removing triglyceride, forming intermediate- and low-density lipoproteins.
In addition to exporting plasma lipoprotein, the liver can oxidize free fatty acids for energy to acetyl coenzyme A (acetyl CoA), a fundamental compound in the tricarboxylic acid cycle (see Figure 16-2). The acetyl CoA thus formed also may be used in the synthesis of other fatty acids, cholesterol, steroids, and ketone bodies, acetoacetate, and β-hydroxybutyrate.5 Furthermore, through the synthesis of acetyl CoA from glucose and most amino acids, the liver is capable of converting carbohydrates and proteins into lipids. Ketone bodies can be exported from the liver and used for energy by peripheral tissues, especially the brain, when glucose is deficient. However, overproduction of ketone bodies can be detrimental, resulting in ketoacidosis.9
Insulin and glucocorticoids closely regulate lipid metabolism6 (see Figure 16-3). Glucocorticoids function primarily to increase fatty acid mobilization from the periphery, whereas insulin decreases adipose tissue release of fatty acids by activating lipoprotein lipase and inhibiting hormone-sensitive lipase. Insulin acts on the liver to increase fatty acid synthesis from glucose.
EXCRETION OF BILE
Bile consists of several components, including conjugated bilirubin, bile acids, cholesterol, lecithin, water, and electrolytes.4 Bile is released by hepatocytes into the bile canaliculi where water diffuses passively. Bile then is transported by large bile ducts and the hepatic duct to the intestine. Water and electrolyte exchange takes place between the bile and the bile duct epithelium; however, isotonicity is maintained. Because the horse does not have a gallbladder or a sphincter at the site of entry of the hepatic duct into the duodenum, the bile is unconcentrated and flow is continuous.1
Bile acids compose 90% of the organic portion of bile.9 Bile acids are amphoteric molecules that act as detergents. These detergents facilitate the excretion of cholesterol and phospholipid from the liver into bile and facilitate the absorption of lipids and lipid-soluble compounds (vitamins A, D, E, and K) from the intestinal tract. The principal primary bile acids (i.e., nondegraded) in the horse are cholate and chenodeoxycholate, both of which are conjugated with taurine.9 Once secreted into the lumen of the intestinal tract, cholate and chenodeoxycholate may be reabsorbed or degraded by bacteria, forming the secondary bile acids, deoxycholate or lithocholate, respectively. More than 95% of the conjugated bile acids excreted in bile and released into the intestinal lumen are reabsorbed by the ileum and returned to the liver via the enterohepatic circulation. Deoxycholate acts as a normal bile acid and can undergo enterohepatic circulation, whereas lithocholate is only reabsorbed once. Bile acids are estimated to be recycled at least 38 times a day in healthy ponies. 9
Bilirubin is the breakdown product of tetrapyrroles that function as electron transport pigments.4 The majority of bilirubin is formed from hemoglobin and myoglobin, but nonheme pigments such as the cytochromes also serve as a source of bilirubin. Macrophages in the spleen, bone marrow, and liver (Kupffer’s cells) engulf the pigments first, convert it to biliverdin (Figure 16-5), and then convert biliverdin to bilirubin and release it from the cell as free, insoluble bilirubin. This form of bilirubin also is referred to as indirect-reacting or unconjugated bilirubin. Unconjugated bilirubin is bound with albumin in the plasma to decrease its hydrophobicity and is delivered to the liver. At the surface of the hepatocyte, the bilirubin is transferred from albumin to ligandin, an intrahepatic transport and storage protein.4,9 Within the hepatocyte, the bilirubin is conjugated with glucuronide in the endoplasmic reticulum. Conjugated bilirubin, also called direct-reacting bilirubin, is water soluble and is excreted into the bile canaliculi. Under normal circumstances, little conjugated bilirubin escapes into the general circulation.
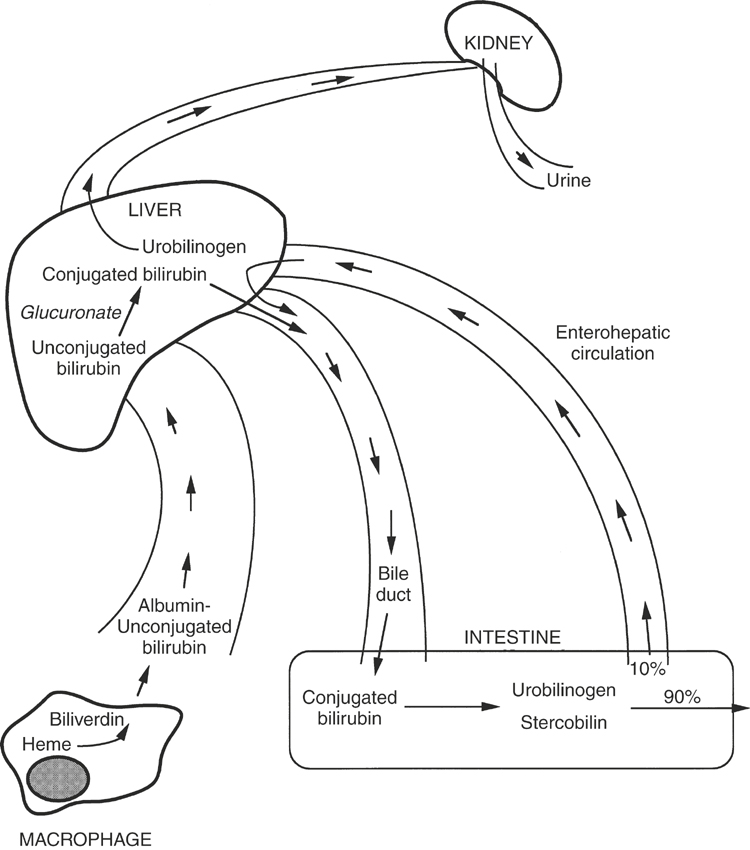
Microflora in the intestinal tract reduce conjugated bilirubin to urobilinogen and stercobilin (see Figure 16-5), which impart a yellow-brown color to feces. In herbivores, the presence of chlorophyll pigments in the feces masks the color of urobilinogen.10 Only in the neonatal herbivore receiving a milk diet are the feces yellow. Urobilinogen is absorbed by the intestinal mucosa and transported back to the liver via the enterohepatic circulation. A small amount of conjugated bilirubin in the intestinal lumen is hydrolyzed to unconjugated bilirubin and subsequently is reabsorbed. The liver extracts most of the urobilinogen; however, a small amount spills over into the urine. Urobilinogen is concentrated in the normally alkaline urine of horses and thus is detectable.10
DETOXIFICATION
The liver is responsible for the biotransformation of numerous endogenous and exogenous compounds. Biotransformation involves a series of enzymatic reactions that alter the physical properties or activity of compounds. Biotransformation occurs in two phases.11 In phase 1, polar groups are added to the compound or existing polar groups are exposed by oxidation, hydroxylation, deamination, or reduction. In phase 2, the product of phase 1 is conjugated, usually with glucuronate or sulfate. Substrates for detoxification usually are water insoluble and biotransformation renders them more susceptible to renal or biliary excretion.4,12 Examples of endogenous substances biotransformed by the liver include ammonia, bilirubin, and steroid hormones (estrogen, cortisol, aldosterone). The liver biotransforms countless exogenous substances, including many drugs, plant toxins, insecticides, and mercaptans.
Phase 1 of biotransformation occurs primarily on the enzyme-bound systems of the endoplasmic reticulum, called microsomes.4,12 Most of these enzymes are iron-containing enzymes of the P-450 system, thus named because they absorb light at 450 nm. The P-450 enzymes are also called mixed function oxidases. Some substrates, referred to as inducers, are capable of saturating the enzymes involved in biotransformation. Enzyme saturation and induction causes hypertrophy of the endoplasmic reticulum and all contained enzymes, thus accelerating substance removal rates. Inducers not only accelerate their own removal rate but also may accelerate the biotransformation of other endogenous and exogenous substances. Examples of enzyme inducers are the barbiturates, phenylbutazone, and chlorinated hydrocarbons. Other agents presented for biotransformation, including chloramphenicol, cimetidine, organophosphates, morphine, and quinidine, may inhibit microsomal enzymes thus prolonging the effect of other substrates. Hepatic biotransformation sometimes results in the formation of a toxic metabolite from a nontoxic parent compound examples being aspirin and halothane.12
MONONUCLEAR PHAGOCYTE SYSTEM
Hepatic macrophages, or Kupffer’s cells, make up a major portion of the mononuclear phagocyte system. Cells of the mononuclear phagocyte system are derived from bone marrow myeloid progenitors and serve two main functions: (1) phagocytosis and (2) to act as antigen-processing cells for lymphocytes. Kupffer’s cells respond to opsonins and synthesize a vast array of inflammatory mediators, including interleukins, tumor necrosis factor (TNF), and eicosanoids. Unlike other macrophages in the mononuclear phagocyte system, Kupffer’s cells function mainly in phagocytosis and are located strategically along the hepatic sinusoids, where portal blood can be cleansed, for example, of bacterial endotoxin, before exposure to the hepatocytes and subsequently the systemic circulation.13 Kupffer’s cells also help cleanse systemic blood entering via the hepatic artery by removing fibrin degradation products (FDPs), tissue plasminogen activators, hemoglobin, microbes, foreign antigens, and other particulate debris.
MISCELLANEOUS FUNCTIONS
The liver serves as a storage site for several vitamins and trace minerals, including vitamins A, D, and B12; copper; and iron. Vitamin D is first converted in the liver to 25-hydroxycholecalciferol and exported to the kidney, where it is transformed into 1,25-dihydroxycholecalciferol, the active form of the vitamin.6 In the fetus the liver is involved in hematopoiesis.4 In the adult the bone marrow serves as the primary site for hematopoiesis; however, the liver may serve as an extramedullary site of hematopoiesis under intense conditions of erythrocyte regeneration or if a large portion of the bone marrow is destroyed.
HEPATIC INSUFFICIENCY
Definition
Hepatic insufficiency or failure refers to the inability of the liver to perform its normal functions properly. Because the liver is involved in such a diverse array of physiologic activities, any pathologic process may hinder one or several functions without impeding others. Furthermore, most hepatic functions are not impaired until greater than 80% of the hepatic mass is lost.4,8,10 The liver also has the capability to regenerate under certain conditions. If hepatocyte loss is gradual and regeneration parallels destruction, then hepatic failure does not necessarily ensue. Thus hepatic disease may be present without accompanying hepatic failure. Consequently, hepatic disease does not always manifest clinically.
Patterns and Pathology of Hepatic Injury
ACUTE FOCAL OR MULTIFOCAL HEPATIC INJURY
Focal hepatic injury occurs with uniform damage to one small area of the liver. Examples of focal hepatic injury include hepatic abscesses, solitary infarctions, and neoplastic growths. Because adequate hepatic reserve exists in the unaffected regions, clinical signs of hepatic failure rarely accompany focal hepatic injury, although evidence of hepatic disease may be demonstrable.11 Acute multifocal hepatic injury is more likely to result in clinically significant hepatic disease. Acute hepatic injury may be degenerative, necrotizing, or inflammatory. Hepatic degeneration refers to a toxic or immunologic insult that causes hepatocytes to swell and take on an edematous appearance.14 Ballooning degeneration is used to describe irregularly clumped cytoplasm with large clear areas.14 If biliary material has been retained, then the hepatocytes appear foamy and swollen (foamy degeneration). Ischemic coagulative necrosis refers to poorly stained and mummified hepatocytes with lysed nuclei, whereas lytic necrosis describes osmotically swollen and ruptured cells.14 Necrosis of contiguous hepatocytes that spans adjacent lobules in a portal to portal, portal to central, or central to central fashion is called bridging necrosis. 14
Hepatic injury may be zonal, that is, affecting certain zones of the liver uniformly throughout the entire organ.11 The liver often appears pale with an enhanced lobular pattern on the cut surface. The two most common types of zonal hepatic injury are centrilobular and periacinar. In centrilobular zonal injury, the area adjacent to the central veins (zone III) is uniformly affected, whereas in periacinar or (paracentral) zonal injury, cellular degeneration involves only a wedge around the central vein (see Figure 16-1). Hepatocytes in these locations are most susceptible to anoxic damage, because the normal oxygen tension is lowest and mixed function oxidase activity is the greatest in these areas. Examples of disease states resulting in centrilobular injury are severe acute anemia, passive congestion caused by congestive heart failure (nutmeg liver), and toxic hepatopathies. Periportal (zone I acinar lobular) injury is rare but may occur with infarction of hepatic vessels, as may occur during verminous arteritis, or exposure to toxins that do not require metabolism by mixed function oxidases (e.g., phosphorus).
ACUTE GENERALIZED HEPATIC INJURY
Acute generalized hepatic injury is often accompanied by clinical signs of hepatic failure, with the extent of damage dictating the severity of the clinical signs.11 Typically, the liver appears pale and enlarged and is often friable. Acute generalized hepatic injury may be the result of infection, necrosis, inflammation, or hepatotoxic agents.4 Bacterial or viral infections, parasitic infestations, or immune disorders may cause acute generalized necrosis or inflammation. Despite its cause, any process that results in an inflammatory response in the hepatic parenchyma is referred to as hepatitis. Acute inflammation most commonly accompanies necrosis and is characterized by the presence of neutrophils and lymphocytes in the areas of cell death or surrounding portal triads. An inflammatory process primarily involving the biliary system is called cholangitis, usually the result of ascending infection from the intestinal tract or after cholestasis.
CHRONIC GENERALIZED HEPATIC INJURY
Chronic hepatic injury is accompanied by clinical signs of hepatic failure when greater than 80% of the hepatic mass is destroyed or replaced by fibrosis.4,8 Fibrosis, the presence of collagen and fibroblasts, occurs when the rate of ongoing necrosis exceeds the rate of regeneration. Typically, the liver appears smaller than normal. Fibrosis commonly follows conditions resulting in chronic hypoxia, chronic inflammation, chronic cholangitis or cholestasis, metastatic neoplasia, trauma, or ingestion of antimitotic agents such as plants containing pyrrolizidine alkaloids. Cirrhosis, or an end-stage liver disease, refers to chronic hepatic disease characterized by the presence of widespread fibrosis, nodular regeneration, and biliary hyperplasia.11 Nodular regeneration, or islands of hepatocytes, occurs when the normal architecture and blood supply of the liver are disrupted or destroyed by the presence of fibrosis. Bridging fibrosis implies fibrosis that extends from one portal area to another or from portal areas to central areas.14 The cause of biliary hyperplasia during chronic liver disease is unknown. One form of chronic hepatic disease, called chronic active hepatitis (CAH), is characterized by the presence of cirrhosis plus an acute inflammatory response.11
Clinical Signs of Hepatic Insufficiency
The clinical signs of hepatic insufficiency are highly variable, nonspecific, and depend on the extent and duration of hepatic disease (Box 16-1). Usually, greater than 80% of the liver mass must be lost before clinical signs become apparent, regardless of the cause of hepatic disease. Thus despite the duration of hepatic disease, the onset of clinical signs is often abrupt. The most common clinical signs of hepatic insufficiency in horses are depression, anorexia, colic, hepatic encephalopathy (HE), weight loss, and icterus.12,15–17 Less commonly reported clinical signs include hepatogenic photosensitization, diarrhea, abdominal pain, bilateral laryngeal paralysis, and hemorrhagic diathesis. Rarely reported clinical signs of hepatic insufficiency in horses are ascites, dependent abdominal edema, steatorrhea, tenesmus, generalized seborrhea, pruritus, endotoxic shock, polydipsia, and hemolysis. The appearance of specific clinical signs of hepatic disease often reflects the type of hepatic function (or functions) that is altered.
HEPATIC ENCEPHALOPATHY
Hepatic encephalopathy (HE) is a complex clinical syndrome characterized by abnormal mental status that accompanies severe hepatic insufficiency.18–20 Clinical signs are widely variable but represent manifestations of augmented neuronal inhibition. This syndrome occurs in patients with advanced decompensated liver disease of all types and may be a feature of acute, subacute, or chronic hepatocellular disease. HE generally is considered to be a potentially reversible metabolic encephalopathy.19 Whether multiple episodes of HE could lead to irreversible neuronal damage is uncertain.
Clinical Signs
No specific features of HE allow this syndrome to be distinguished from other causes of cerebral dysfunction. The earliest phase of HE is probably missed in most equine patients because it represents minimal behavioral changes with subtle impairment of intellect because of bilateral forebrain dysfunction21 (stage I; Table 16-1). In humans, these early signs are more apparent to close friends and family members than to a physician. As encephalopathy progresses, motor function, intellectual abilities, and consciousness become impaired; generally at this stage (corresponding to stage II) horses become obviously affected. Clinical signs include depression, head pressing, circling, mild ataxia, aimless walking, persistent yawning, and other manifestations of inappropriate behavior. Somnolence develops. Next the horse is rousable but responds minimally or excessively to the usual stimuli. At this stage (III) the horse often manifests aggressive or violent behavior interspersed with periods of stupor. Finally, consciousness fades, the horse becomes recumbent, and coma ensues. Occasionally seizures occur during the later stages of HE, but in general they are atypical. The severity of encephalopathy corresponds to the degree of hepatic dysfunction; however, neither of these parameters correlates with type or reversibility of the underlying hepatic disease.
TABLE 16-1 Clinical Stages of Hepatic Encephalopathy
| Stage | Mental Status |
|---|---|
| I | Mild confusion, decreased attention, slowed ability to perform mental tasks, irritability |
| II | Drowsiness, lethargy, obvious personality changes, inappropriate behavior, disorientation |
| III | Somnolent but rousable, marked confusion, amnesia, occasional aggressive uncontrolled behavior |
| IV | Coma |
Adapted from Gammel SH, Jones EA: Hepatic encephalopathy, Med Clin North Am 73:793–813, 1989.
Cause and Pathophysiology
By definition, the cause of HE is insufficient hepatocellular function, irrespective of the cause of the liver disease. That is, a normally functioning liver is necessary to maintain normal brain neuron and astrocyte function. With acute liver disease, HE is primarily caused by astrocyte swelling, acute cytotoxic cerebral edema, and intracranial hypertension. In chronic hepatic disease, HE develops more insidiously. Astrocytes are swollen but also show evidence of Alzheimer type II changes.22 Whether acute or chronic in nature, the precise pathogenesis of HE remains unclear, and considering the numerous proposed hypotheses, the cause is almost certainly multifactorial. The following mechanisms have been suggested for the development of HE and any or all may be involved to greater or lesser degree:
Perhaps the oldest and most “predominant” hypothesis for HE involves the accumulation of toxic materials in the blood (derived from the metabolism of nitrogenous substrates in the gastrointestinal tract) that bypass the liver through functional or anatomic shunts.18,21,23 Accordingly, HE may be caused primarily by failure of the liver to adequately remove certain substances from the blood that have the direct or indirect ability to modulate function of the CNS. Ammonia, after the degradation of amino acids, amines, and purines by enteric bacteria, has been supported widely as a major neurotoxin of hepatic disease.22,23–25 In patients with liver failure, ammonia is insufficiently metabolized through the urea cycle; thus plasma concentrations increase, and ammonia enters the CNS, where it may cause encephalopathy.23,26 Evidence also points to zinc deficiency, which is important for the urea cycle, in the pathogenesis of HE.27
Ammonia has a toxic effect on cell membrane neurons by inhibition of the Na,K-dependent adenosine triphosphatase activity in nerve cell membranes, causing depletion of adenosine triphosphate.25,27 Hyperammonemia also is associated with a disturbance in CNS energy production caused by alterations in the tricarboxylic acid cycle that result in a decrease in α-ketoglutarate formation and increased synthesis of glutamine.28 Astrocytes in the brain also detoxify ammonia by synthesizing glutamine through amidation of glutamate. Glutamine accumulation in astrocytes is a major cause of cell swelling and generation of cerebral edema in acute fulminate hepatic failure.22,27
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


