(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
I am an undergraduate teacher and that is what my first love has been. Over the years, learning from others as well as by my own experience, I could figure out that the best method to teach concepts in science to the young minds is the method of doubt and challenge. Doubting and challenging everything that a textbook has or a class teacher says pay large dividends. This way, students learn inferential logic, nature of evidence, experimental designs, hypothesis testing, alternative interpretations, resolving between alternative interpretations, and the required openness of mind to do so in a much more robust and sound way than the conventional “textbook as bible” approach. In the doubt and challenge approach, students are encouraged to contest prevalent concepts and come up with alternative interpretations of the same experiments and data on which the textbook concepts are based. Further, if any of their alternative interpretations look promising even at a speculative level, they are encouraged to suggest experiments that could distinguish between the conventional and alternative explanations of the phenomenon. In order to satisfy ourselves we then have to go beyond textbooks and explore what is happening in that field currently, see whether the experiments that we visualized have been done or are being done anywhere and, if so, what they imply. Most often, the textbook “wins,” but not uncommonly we identify significant gaps in the knowledge, some logical inconsistencies that need to be addressed, many unanswered questions, and more frequently many unquestioned answers! It is just too common to find that although the frontiers of research are expanding rapidly, some simple fundamental questions remain not only unanswered but knowingly or unknowingly trivialized and ignored.
I am an undergraduate teacher and that is what my first love has been. Over the years, learning from others as well as by my own experience, I could figure out that the best method to teach concepts in science to the young minds is the method of doubt and challenge. Doubting and challenging everything that a textbook has or a class teacher says pay large dividends. This way, students learn inferential logic, nature of evidence, experimental designs, hypothesis testing, alternative interpretations, resolving between alternative interpretations, and the required openness of mind to do so in a much more robust and sound way than the conventional “textbook as bible” approach. In the doubt and challenge approach, students are encouraged to contest prevalent concepts and come up with alternative interpretations of the same experiments and data on which the textbook concepts are based. Further, if any of their alternative interpretations look promising even at a speculative level, they are encouraged to suggest experiments that could distinguish between the conventional and alternative explanations of the phenomenon. In order to satisfy ourselves we then have to go beyond textbooks and explore what is happening in that field currently, see whether the experiments that we visualized have been done or are being done anywhere and, if so, what they imply. Most often, the textbook “wins,” but not uncommonly we identify significant gaps in the knowledge, some logical inconsistencies that need to be addressed, many unanswered questions, and more frequently many unquestioned answers! It is just too common to find that although the frontiers of research are expanding rapidly, some simple fundamental questions remain not only unanswered but knowingly or unknowingly trivialized and ignored.
The method of doubt and challenge used in undergraduate class has shaped my thinking permanently in such a way that when I got interested in the evolutionary origins of diabetes and related disorders, I found myself extensively using the method of doubt and challenge while trying to read basic textbooks on physiology and pathology of diabetes. Here I was playing the teacher as well as the student myself. Very soon I found myself surprised and amazed by the number of logical inconsistencies and paradoxes present in this field. For any teacher using the method of doubt, it is not uncommon to find some or the other problem in every textbook chapter. But the problems with diabetes appear to be unmatched both qualitatively and quantitatively. Fortunately or unfortunately, I never formally taught the physiology or pathology of diabetes to any class any time. I will try to do that in this chapter now playing both student and teacher myself. As in the classroom, the basic intention here is to develop an insightful understanding. Critical thinking reinforces concepts that are quite sound, refines the ones that are not so much, and makes us rethink about the ones that are not. Only in rare cases the classroom method of doubt turns something upside down. The teacher in me is generally convinced that the textbook is correct most of the times and therefore defends the textbook, whereas the student in me is skeptical. Let us look at the popular textbook portrait of type 2 diabetes from the point of view of this student–teacher duo.
We can best begin by looking at the five classical postulates of T2D based on the textbook picture. Much research in the last two decades has indicated the necessity to drift away from the classical picture, but in order to understand the necessity to drift, we need to take a critical look at the classical picture first:
1.
Obesity is a consequence of positive energy balance, that is, the net energy intake being greater than the net energy expenditure.
2.
Obesity is the main cause of insulin resistance.
3.
The body tries to compensate insulin resistance by making the pancreatic β cells produce more insulin.
4.
There is a progressive degeneration of β cells presumably due to “exhaustion” resulting in relative insulin deficiency, that is, insulin levels being short of compensating for insulin resistance. A combination of insulin resistance and relative insulin insufficiency (IR–RII) results in hyperglycemia.
5.
The chronically increased blood sugar levels lead to all other pathophysiological consequences of diabetes.
The sequence appears very logical and convincing at a glance. We have seen this in moderate details in the last chapter. Now, we will start looking at the detailed mechanisms and evidence using the method of doubt and challenge in the hypothetical classroom.
1.Energy balance and obesity: The baseline assumption of obesity research is the energy balance equation. It simply says that if the net energy intake of the body is greater than the net energy consumption, body mass will increase and such a continued imbalance over a sufficiently long time will lead to obesity [1]. The equation is nothing but fundamental thermodynamics and therefore cannot be doubted. What the skeptic student wonders is not whether the energy balance equation is right or wrong, but whether it says anything useful about obesity. It is like answering the question “why did you fail in the exams?” by saying “because I got less than the passing marks.” This is only saying the same thing in different words, not reasoning out. So the student is not convinced that the energy balance equation “explains” obesity. The student’s dissatisfaction is not new. Inadequacy of the thermodynamic energy balance in explaining obesity is recognized from time to time [2, 3]. The teacher, however, knows that the usefulness of the energy balance equation lies in defining the question in more objective terms, not in answering the question. Therefore it is not good to be satisfied by the energy balance equation but at the same time not prudent to discard it. The balance equation forms the platform on which real questioning can begin. In modern lifestyle, food intake has increased, and physical activity has come down resulting in a positive shift in the energy balance. This sounds temptingly simple and logical to believe. In literature, there appears to be no agreement on whether it is increased food intake or decreased activity that is primarily responsible for obesity [4]. The available evidence is contradictory. Experiments with doubly labeled water that measure the total energy expenditure of the body have demonstrated that modern lifestyle has not changed the mean total energy expenditure sufficiently to account for the obesity epidemic [5, 6]. This implies that our energy intake must have gone up. The increased fat content of food is often blamed to be responsible for obesity. This is the most dominating line of thinking in the antiobesity drive. The drive for low-fat diet has resulted in a rapidly growing market for low-fat foods in America, for example, but the increased consumption of low-fat foods has not resulted in curbing obesity [7]. In stark contrast lies the example of the East African Masai tribe whose normal diet has a high proportion of animal fat (60–70% of total energy intake), but obesity, insulin resistance, and hypercholesterolemia are extremely rare among them [8–10].
Perhaps, the teacher tries to defend; in the example of the Masai tribesmen, the energy expenditure is more important than energy intake. The importance of gluttony and sloth could be different in different populations leading to contradictory results across studies. But whether increased energy consumption or decreased expenditure is responsible for the obesity epidemic is not the most important question. Let us assume that both are happening simultaneously resulting in positive energy balance. The real question is what makes the positive energy balance possible when the body has a wide variety of mechanisms that regulate energy intake. Do we eat more than what our body requires only because more is available? Is our energy intake regulated only by how much is available? There is no doubt that the availability of food has increased in many modern human societies [11], and that is believed to be the sole reason for the obesity epidemic by some researchers [12]. The belief that food availability alone drives obesity contradicts the large body of evidence for a variety of elaborate mechanisms of energy homeostasis regulating food intake. A large number of different mechanisms of the body regulate food intake as well as fat storage [13–16]. Signals for regulation for food intake are generated starting from the taste buds and continuing to further levels including distension of the stomach, chemical signals in the upper and lower parts of the gastrointestinal tract, blood levels of sugars and amino acids, insulin signaling, leptin–adiponectin and other signals by adipose tissue, glucose sensing in the portal vein, and glucose sensing neurons in the brain that lead to neuronal circuits regulating food intake (Table 3.1).
Table 3.1
Mechanisms of food intake regulation
Signal | Site of production | Stimulus | Site of action | Action on appetite | |
|---|---|---|---|---|---|
Gut and pancreatic signals (short-term regulation) | |||||
1 | Sensory | Oropharynx | Food taste, swallowing | ? | ↓ |
2 | Physical | Stomach | Distension of stomach | ? | ↓ |
3 | Insulin | Pancreas | Plasma glucose | ARC, DMH, PVN | ↓ |
4 | Pancreatic polypeptide (PP) | Pancreas | Food intake | Area postrema | ↓ |
5 | PYY | L cells of GIT | Food intake, particularly fat | Peripheral, arcuate NPY neurons | ↓ |
6 | Ghrelin | Gastric oxyntic cells, other parts of GIT | Circadian, stimulated by food anticipation, suppressed by food intake | Hypothalamic ARC | ↑ |
7 | GLP-1 | L cells of intestine | Food intake | Pancreas, CNS | ↓ |
8 | Oxyntomodulin (OXM) | L cells of intestine | Circadian, food intake | Hypothalamus | ↓ |
9 | Cholecystokinin (CCK) | Duodenum and jejunum of GIT, CNS | Food intake, aggression, anxiety | CNS—NTS, DMH, area postrema | ↓ |
10 | Bombesin | Gut | Food intake | CNS | ↓ |
Adipocity signals (long-term regulation) | |||||
11 | Leptin | Adipose tissue | Increased adiposity | POMC, CART neurons | ↓ |
12 | Adiponectin | Adipose tissue | ? | Hypothalamus, skeletal muscle | ↓, increased energy expenditure |
Central nervous system | |||||
13 | Neuropeptide Y | ARC | Fasting | Hypothalamus | ↑ |
14 | Melanocortin–POMC products | Hypothalamus | Food intake, insulin, leptin | Hypothalamus—ARC, VMH, and PVN | ↓ |
15 | Melanocortin, Agouti, and AgRP | Peripheral, CNS | Fasting | ARC | ↑ |
16 | CART | ARC, LHA, PVN | Leptin, fear–anxiety | Hypothalamus | ↓ |
17 | Serotonin | Brain | Food and other satiation responses | ARC | ↓ |
18 | Glucose sensing neurons | Hypothalamus and other parts of brain | Glucose levels in the brain | Hypothalamus | ↓↑ |
19 | Reward circuits opioid, endocannabinoid, dopamine | Brain | Food and other “rewards” | Hypothalamus NAc | ↓↑specifically high-energy food |
History of experimental investigations on food intake control is quite old and interesting. In some of the early rat experiments where there was an open esophageal fistula that drained out the food taken in so that no food entered the stomach, the animals stopped eating after a certain meal size. This indicates that some food intake regulation mechanisms exist even before the food reaches the stomach [17]. The meal size in such sham feeding trials is somewhat larger and the inter-meal interval shorter, indicating that sham feeding is unable to induce the same satiety response as that obtained by a distended stomach [17–19]. Nevertheless some regulation does work at the oropharynx level too and is influenced by and interacts with other food-related hormonal signals such as insulin and cholecystokinin (CCK) as well as neuronal processes [17, 20–23]. The stomach level of control was shown to work independent of taste buds. In these experiments, rats were fed liquid diet through a tube bypassing the oropharynx. The animals themselves controlled food intake by pressing a lever for which they were adequately trained. Although these animals had easy, automated, and unlimited access to food, they did not grow obese [24], demonstrating that easy availability of food was not enough. Experiments that excluded both oral and gastric influences and directly introduced food in the jejunum via a fistula demonstrated that regulation mechanisms exist at the level of the intestine independent of oral and gastric signals [25]. This demonstrates that different levels of food intake regulation are at least partly independent of each other and one can function in the absence of the other. Today a large number of molecular and neuronal mechanisms of regulation of energy intake are known that act at acute or chronic level (Table 3.1). It is difficult to perceive how such a system with a series of backups has no role and only the availability of food decides food intake. Therefore there must be something beyond greater availability of food that is responsible for increased food intake.
In the context of the modern obesity epidemic, one should be able to explain why the regulation mechanisms appear to be failing today. Can it be explained by any defect in the regulation mechanisms? Since the energy homeostasis mechanisms are multileveled and independent, if one system fails, others are there to compensate. Therefore a single defect is most unlikely to cause overeating. This is indeed true since the total number of cases in which obesity could be traced back to a single genetic defect is surprisingly small (about 200) [26] in the entire history of medicine and human genetics. On the one hand, the system is difficult to get perturbed by any single defect, and on the other it is most unlikely that many or most of the regulation mechanisms will develop a mutation or an acquired defect simultaneously in a large proportion of the population. Therefore such a large fraction of the population getting obese in spite of the presence of multilevel food regulation mechanisms that evolved and worked well over millennia is an unresolved riddle. Increased availability and security of food is not a sufficient cause. In case it is, one still needs to explain why the regulation mechanisms fail.
A large number of studies have focused on the molecular mechanisms of energy homeostasis, and many of them hope to develop an effective antiobesity drug [16]. There is a major missing link in the whole of this research. Energy homeostasis is not the only thing that regulates food intake. There are many ecological, social, and behavioral factors that are equally important determinants of eating behavior. For example, after a successful hunt, a mother cheetah may not eat in spite of being hungry if feeding the cubs is a priority. A number of animals store and/or hide food. Such an animal would recover the cached food when hungry. However, it may refrain from doing so if another strong and dominant competitor is watching since there is a risk of the food being snatched away. Foraging for food may increase risk of predation, and an animal may choose to stop foraging and eating at much lower levels of satiety in the presence of a predator than in its absence. Not much research has addressed how these behavioral signals interact with the energy homeostasis signals at the molecular and neuronal level. This appears to be due to a tradition in which biochemists generally do not think of behavior as a prime driver of biochemical regulation. As a result, the mainstream thinking appears to have stuck at the narrow vision of energy homeostasis and energy balance equation. The possible effects of behavioral inputs in metabolic regulation have not been adequately considered. But there is a reason to suspect that obesity may be primarily a result of a change in the behavioral components rather than energy homeostasis mechanisms. In the last few decades in which obesity attained epidemic proportions, human ecology, social structure, and behavior have changed substantially, whereas there is no apparent reason why the biological mechanisms of energy homeostasis that have evolved and worked for thousands of generations may suddenly have changed in such a large fraction of the population in a short time. Therefore there is a need to seriously rethink the energy balance and energy homeostasis paradigm. The roots of obesity may lie in social structure and behavior rather than diet. This needs alternative thinking, and we will reserve it for another chapter ahead. For the time being, let us continue to critically examine the textbook picture.
The current thinking about diet composition is closely linked to the obesity centered view. Therefore it would be appropriate to consider it here itself. A number of studies focus on dietary fat rather than accumulated fat as an inducer of insulin resistance [27–29]. Whether high-fat diet or high-carbohydrate diet is linked to obesity and insulin resistance syndrome is highly debated. There has been a fat school and a carbohydrate school, and both have some data in support and possible mechanisms to explain the contribution to obesity [30–40]. A less known possibility is that even high-protein diet would promote insulin resistance. This is logically very sound because the brain needs glucose as the source of energy and it cannot utilize fatty acids or amino acids catabolically. When dietary carbohydrate intake is negligible, the liver needs to make sufficient glucose for the brain and insulin resistance promotes liver gluconeogenesis. Therefore high-protein, low-carbohydrate diet should also induce insulin resistance. Amino acids stimulate mTOR activity [41], and mTOR appears to induce insulin resistance [42]. Thus a mechanism by which high-protein diet can potentially induce insulin resistance also exists. There is an odd piece of evidence in support of it too. Insulin resistance is demonstrated in dolphins under high-protein diet [43]. In humans, high-protein diets do not seem to be generally associated with insulin resistance, but protein deprivation has been reproducibly shown to increase insulin sensitivity, the effect being independent of total calorie intake [44–46]. Therefore some positive association between protein intake and insulin resistance can be shown in humans too. There has been an interesting suggestion that ethnic groups that historically depended more on protein-rich diet evolved a tendency towards insulin resistance (McMichel 2001, source reference [47]). The point I want to emphasize here is that there can be arguments and evidence for high-fat, high-carbohydrate, or even high-protein diet causing insulin resistance. Literature on diet and insulin resistance is full of internal contradictions. One would find some evidence for high-fat or high-carbohydrate or high-protein diet causing insulin resistance and also would find sufficient evidence against. Perhaps the effects of dietary composition are non-monotonic and an optimum balance between protein, fat, and carbohydrate could be important, and such recommended ideal composition has been prescribed [48, 49]. Perhaps a balanced diet is the key and loss of balance in any direction could lead to insulin resistance. But while talking about balanced diet, one should not forget what the diversity of people across the globe have been consuming traditionally without developing obesity, T2D, and related disorders on an epidemiological scale.
Even a quick glance at the traditional dietary compositions of different tribes in the world reveals that people have adapted to different extremes of diets under different ecological settings. There are examples of tribes taking over 85% carbohydrates, 70% animal fat, or almost 100% dependence on meat and fish with practically no carbohydrate intake (Fig. 3.1). As compared to the wide variation in diet on which these different societies have survived without significant incidence of the modern lifestyle disorders [50, 66], the dietary changes witnessed by the last few generations in the modern Westernized societies are meager. But there lies the paradox of diet composition. Within the Westernized populations, there is good evidence that high-fat or high-soluble carbohydrate diet causes obesity or insulin resistance [51, 52, 67]. But much more extreme fat- or carbohydrate-consuming tribes do not show obesity and metabolic syndrome [10, 32]. The Masai derive 60–70% energy from animal fat; Kalahari !Kung derive almost the same from plant lipids. West Africans and traditional Pima Indians had a predominantly carbohydrate diet, and Eskimos’ traditional diet had hardly any carbohydrates. It may be argued that since these tribes are on extreme diets for several generations, they may have evolved genetic resistance against diet-induced obesity. But this is not true since on adopting modern Western lifestyle, they rapidly develop signs of metabolic syndrome [68–71]. Today the macronutrient composition of the diets in America, where the prevalence of obesity is the highest, or India that has highest number of diabetics is much more “balanced” than these extreme diet communities (Fig. 3.1). But interestingly, the extreme diet communities traditionally had negligibly small prevalence of diabetes, hypertension, or high cholesterol. This does not imply that they have no health problems. The point relevant here is that their health problems are not diabetes, hypertension, and cardiovascular diseases (CVD), as long as they remain in their traditional lifestyle. This suggests that there might be something other than dietary macronutrient composition in the process of acculturation to modern urban lifestyle that is crucial [72]. Normally the human body appears to have the capacity to adapt to widely different dietary compositions.
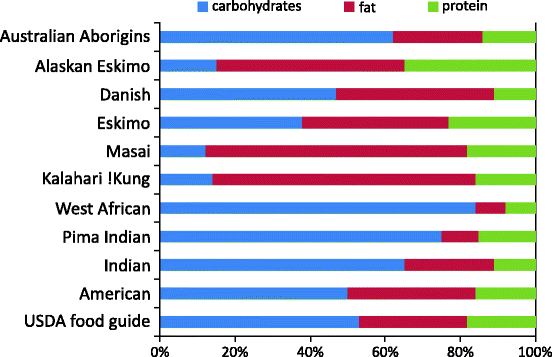
Fig. 3.1
Macronutrient composition of traditional and modern diets around the globe. Bars denote the percentage of energy obtained from carbohydrates, fats, and proteins, respectively. It can be noted that average diet composition in America (one of the countries with the highest incidence of obesity) and India (one of the countries with the highest number of type 2 diabetics) is nearest to the prescribed ideal composition. Many other societies with more extreme diets have much lower incidence of obesity, diabetes, and related disorders. Data compiled from multiple sources [32, 50–65]
If we believe that the dietary macronutrient composition drives obesity, the student has another fundamental question which needs an answer. With the exception of essential amino acids and certain fatty acids that our body cannot synthesize, carbohydrates, fats, and proteins are interconvertible in the body, and mechanisms exist to convert carbohydrates into fat or protein and vice versa. These mechanisms are activated according to the metabolic demand, and metabolic demands are largely decided by behavior. A wrestler can build up sufficient muscle protein, even if the diet is predominantly carbohydrates. There are many examples of vegan body builders, wrestlers, and athletes whose total protein intake is substantially less than meat eaters. Masai with high-animal fat diet are lean, insulin sensitive, and having low cholesterol levels [8–10]. Kalahari !Kung that consume high proportion of plant lipids were found to have a marginally impaired glucose tolerance by the Western standards. But this was not due to insulin resistance since their insulin resistance index (HOMA-IR) was invariably low [73].
The real importance of diet composition is in supplying molecules that our body cannot synthesize including vitamins, minerals, essential amino acids, and molecules such as omega 3 fatty acids. Having supplied these according to the requirement, whether energy comes in the form of fat, protein, or carbohydrate should not matter too much since the body can derive energy from all of them. Therefore the relative proportions of carbohydrate, fat, and protein are unlikely to be central to obesity. If negative feedback-based mechanisms exist for regulation of food intake, even the energy density of food should not matter. An energy-dense food may escape short-term regulation mechanisms such as signals given by distension of stomach but is unlikely to escape long-term regulation mechanisms driven by leptin and other adipose tissue signals, provided such mechanisms work. If they do not, the reason for their failure needs to be blamed and not the energy density. The critical factor therefore is not the nature of food but the apparent failure of energy intake regulation mechanisms.
Thus there are problems with all the three lines of mainstream thinking. If we believe that reduced energy expenditure is the main cause, doubly labeled water studies are not in support. If we think that increased energy intake is central to obesity, we have not yet answered why energy intake regulation mechanisms fail. If dietary composition is to be blamed, global anthropological data are not in support. This means that we really do not know the predominant cause of obesity as yet, at least not at a depth sufficient to satisfy students in my undergraduate classroom. Some alternative ways of thinking are coming up recently which we will elaborate subsequently. At this point, I need to keep the discussion on the origins of obesity quite brief for two reasons. One is that sufficient critical literature is already there examining this question (e.g., [33, 47, 74]), and the other is that the origin of obesity is not the prime question of interest here. Two other questions are of greater concern and inadequately addressed so far, namely, whether obesity is central to insulin resistance and whether insulin resistance is central to T2D as generally believed. These two need to be looked at with much more rigor and critical questioning.
2.Obesity as the cause of insulin resistance: There is a strong body of evidence that obesity is correlated to insulin resistance. However there is much ambiguity about whether obesity causes insulin resistance or insulin resistance leads to obesity. James Neel whose thinking has been central to the prevalent concept of thrifty gene [75] did not think that obesity led to diabetes. He rather stated that a “diabetic tendency” (the concept of insulin resistance was not there when Neel wrote this paper) was responsible for obesity. If the “diabetic tendency” is insulin resistance, then insulin resistance should be the cause of obesity. There is some evidence that insulin resistance could be the cause rather than the effect of obesity. Muscle-specific insulin receptor knockout mice become obese [76–78] demonstrating that induced muscle insulin resistance can lead to fat accumulation provided fat cells remain insulin sensitive. Fat cell-specific insulin resistance, on the other hand, arrests fat accumulation [77, 78]. It is possible therefore that if muscle cells become more insulin resistant than fat cells, then that specific pattern of resistance can lead to obesity. However, whether fat cells remain relatively insulin sensitive in an insulin-resistant individual has not been clearly shown. Other possible mechanisms have also been suggested through which not insulin resistance per se but hyperinsulinemia leads to obesity [79, 80]. This is a part of a different line of thinking that we will see in sufficient depth later.
The current mainstream thinking appears to go in the other direction. Obesity is perceived to be the major clinically important cause of insulin resistance by the current perception. Doubts can be raised whether obesity is the sole factor leading to insulin resistance since a significant proportion of type 2 diabetics or insulin-resistant persons are of normal weight [81–83]. In India, for example, it is just too common to find normal weight or even lean and thin type 2 diabetics. Also obesity is not related to cardiovascular mortality in India as it is in Western countries or Eastern Asia [84]. The teacher now defends the textbook by showing that although the normal weight diabetic individuals may not have a high BMI, they have a higher proportion of body fat and that is what makes them insulin resistant. Here again, the more important question appears to be that if the net energy balance is not positive (as indicated by normal BMI), what leads to higher fat percentage in such individuals? It needs to be appreciated that this is a different question. It is not an energy balance question. If the proportion of fat, not body weight, is what counts, we need to think on a different line. This distinction is often not very clearly made. Body composition is decided by factors other than energy balance, and these factors are not yet clearly known.
The other side of the coin is that not all obese individuals are insulin resistant [83, 85]. This is defended by the teacher saying that it is the individual genetic makeup that decides this. Just as everyone does not get cold when a common cold virus is around, obesity does not lead to insulin resistance in every case. But while saying this, the teacher in me knows that this is a lame argument. We will see in the following chapter all the new evidence (or lack of it?) about the role of genes in obesity and diabetes to realize that after the genome data became available, the genetics argument has almost completely lost ground. But there is another possible argument that might come handy in defense of the obesity school. It is not the body mass index (BMI) or total fat that is so much important. There is evidence that it is central obesity or visceral abdominal fat which induces insulin resistance. Particularly interesting is the case of sumo wrestlers who have a high BMI, a large amount of body fat, and abdominal obesity as well. But as compared to other individuals of equal BMI or equal waist circumference, they are much less insulin resistant as long as they are active wrestlers [86]. So their abdominal obesity does not appear to have the same effect as in other obese individuals. This is defended now by showing that sumo wrestlers have more subcutaneous fat and little visceral fat [87] and therefore they are less insulin resistant. Subcutaneous fat appears to be relatively harmless. There is some evidence in support of this argument [88–90]. The next natural question is exactly what makes the fat stores of sumo wrestlers go subcutaneous and not visceral. Again the energy balance equation is useless to answer this question. Neither total energy intake nor expenditure can explain the distribution of fat in the body. Therefore there must be something else that decides the distribution of fat, and this something else must be more important than energy intake and expenditure. Currently there is no satisfactory answer to what this “something else” is.
Is the phenotypic association between obesity and insulin resistance reflected at the level of genes as well? A large number of alleles have been shown to have statistically significant associations with obesity and a number of alleles with T2D although the effect size is very small. Out of the recently identified risk alleles for T2D, very few are related to obesity parameters [91]. A locus near IRS1 associated with lower body fat percentage is associated with higher insulin resistance, adverse lipid profile, and low adiponectin in men [92], indicating that the association between obesity and insulin resistance is not obligate at the genetic level.
Nevertheless, let us assume that the association between obesity and insulin resistance is real and causal. The next question is: What is the mechanism by which obesity induces insulin resistance? We have seen in the last chapter that a large number of potential mechanisms have been suggested, but we do not know with sufficient clarity which of them plays a significant role in the current obesity epidemic. Whether FFAs are the main players or adipokines or both is not very clear, and the undergraduate class demands this clarity.
For a long time, circulating lipids and free fatty acids are considered responsible for inducing insulin resistance. However the mechanism by which lipids or FFAs induce muscle insulin resistance is still elusive.A number of hypotheses have been floated, but none has been convincingly demonstrated. For example, one of the earlier hypotheses called Randle hypothesis speculated that fatty acids competed with glucose for substrate oxidation in muscle leading to increased intracellular glucose-6-phosphate [93, 94]. This resulted in accumulation of intracellular glucose and thereby reduced further uptake of glucose by muscle. However later studies in normal individuals with increased fatty acid levels showed that there is actually a fall in the intracellular glucose-6-phosphate and glucose levels. This fails to support the Randle hypothesis. Another hypothesis is based on the observed dysfunction of muscle mitochondria in insulin resistance. Based on correlative evidence, it was proposed that lipid overloading of muscles leads to incomplete fat oxidation by mitochondria. The resultant defective mitochondrial function underlies insulin resistance. However, experiments attempting a direct test of the hypothesis found that lipid accumulation actually increased mitochondrial proteins and their function [95]. If lipids were responsible for reduced mitochondrial function, lowering of lipids should have increased it. However, pharmacological lowering of plasma lipids in insulin-resistant subjects reduced mitochondrial gene expression instead of increasing it [96]. Also demonstration of mitochondrial dysfunction in non-obese first-degree relatives of diabetics [97] suggests that the mitochondrial problem appears to be due to “something else” and is not fat induced.
Another strong line of argument against FFAs being responsible for muscle insulin resistance is based on the measurement of insulin resistance by hyperinsulinemic euglycemic clamp method. In this method, continuous insulin infusion is done to raise and maintain a stable steady high level of insulin in blood. The stable hyperinsulinemia thus achieved rapidly depletes FFAs as has been clearly demonstrated. If standing levels of FFAs were responsible for insulin resistance, then insulin resistance should not be seen in a hyperinsulinemic euglycemic clamp study. But the clamp method is known to measure insulin resistance highly reproducibly, and insulin resistance thus measured matches very well with other methods that work in the physiological range of insulin and thereby not depleting FFAs. If depleting FFAs does not reduce insulin resistance, at least the standing FFA levels cannot be said to be responsible for insulin resistance.
As the mechanisms by which FFAs or circulating lipids induce insulin resistance are problematic, there is a growing body of evidence supporting the role of adipokines in inducing insulin resistance. Adipokines are secreted by adipose tissue, and therefore, the focus shifts from circulating FFAs and lipids to stored lipids. The truth most inconvenient for this thinking is that if adipocytes secrete proinflammatory and pro-insulin-resistance signals, the same adipocytes also secrete anti-inflammatory and insulin sensitizing signals. Adiponectin is an important signal molecule secreted by adipocytes which has anti-inflammatory, antiobesity, and insulin sensitizing action [98–101] and which is paradoxically under-expressed in obesity although the tissue secreting it is increased in volume. Another adipocyte-secreted protein is sFRP5 which has anti-inflammatory and antidiabetic effects [102]. Normally leptin produced by adipose tissue regulates energy intake and thereby arrests obesity. In healthy life, the adipose tissue signals are bidirectional, and these opposing signals could be balancing out quite well, but in obesity-induced disorders, the proinflammatory and pro-insulin-resistance signals predominate and their counterpart gets suppressed. So it is not the amount of adipose tissue per se but the shift in the balance of the proinflammatory and anti-inflammatory signals given by the adipose tissue is what appears to be important. A systems biology approach to diabetes in mice has demonstrated that proinflammatory adipokine secretion is detectably greater in diabetes-prone than diabetes-resistant mice at 6 weeks of age when there is no detectable difference in the fat mass of the two. With age and fat feeding, the inflammatory response in diabetes-prone mice increased along with obesity, but that in diabetes-resistant mice did not [103]. This suggests that the shift in balance of proinflammatory and anti-inflammatory actions of adipose tissue might be happening first and obesity arises subsequently. What causes this shift in balance is naturally the question that follows, and there is no textbook answer to it mainly because this question appears to be hardly ever asked before. Again the answer must lie in “something else” and not in obesity per se.
Particularly intriguing is the fact that adiponectin, a molecule that is normally synthesized by adipose tissue in large quantities, is negatively associated with obesity. If it is secreted by adipose tissue, the greater the adipose tissue, the greater should have been the secretion, but in reality it is correlated negatively with obesity. This paradox is well recognized but so far there is no solution. The negative correlation of adiponectin with obesity and the antiobesity action of adiponectin [104, 105] together imply that adiponectin deficiency is more likely to be a cause rather than an effect of obesity. Both logic and evidence support the view that the shift in the balance of adipokines should be arising first and increased fat mass a consequence of it. Since adipocytes produce leptin which regulates food intake and adipogenesis [106, 107], and adiponectin and sFRP5 which arrest fat accumulation, obesity is not possible unless leptin resistance [108] develops and a deficiency of adiponectin and sFRP5 develops. This is not a hen and egg problem. “Obesity first” is just not logical; leptin resistance and adiponectin–sRFP5 deficiency have to arise first, and only then is obesity possible. The shift in balance of adipocyte secretions must clearly precede obesity, and what shifts the balance should be a matter of greater concern than obesity itself. But this question does not have logical solutions so far. This is the major weakness in the prevalent thinking that adipose tissue induces insulin resistance through adipokines. Again we need to bring in that mystic “something else” that changes the adipokines balance. Obesity is made possible by this shift in adipokine balance.
If we assume that this paradox is somehow resolved and satisfy ourselves that the anti-inflammatory adipokines are somehow suppressed and the proinflammatory adipokines have a crucial role in inducing insulin resistance, another contradiction awaits us. Anti-inflammatory steroids such as cortisol should antagonize obesity-induced insulin resistance owing to their anti-inflammatory action. But the reality is diametrically opposite. The anti-inflammatory cortisol has a worsening rather than improving effect on insulin resistance [109]. Therefore assuming inflammatory mediators responsible for the loss of insulin sensitivity is not free of problems.
A different hypothesis linking fat to insulin resistance is adipose tissue expandability hypothesis [85, 110]. It postulates that fat stored in regular adipose depots is not harmful. But when these depots have reached their capacity, there is accumulation of ectopic fat, that is, fat is stored in tissues where it is not normally stored. For example, lipids stored in muscle, called intramuscular triglycerides (IMTG), are said to cause insulin resistance, and this is supported by the observation that insulin-resistant individuals have high IMTG [111–115]. Regarding the role of IMTG, there is a paradox. Athletes are typically rich in intramuscular fat and that is adaptive for them. Regular endurance exercise appears to increase IMTG [116]. In spite of high IMTG, athlete muscles are highly insulin sensitive demonstrating that lipids could not be primarily responsible for muscle insulin resistance [117–119]. The correlation between IMTG and insulin resistance is funny. If only non-exercising individuals are considered, there is a significant positive correlation between IMTG and insulin resistance. If athletes are added to this sample, the correlation gets destroyed since they have high IMTG and still are insulin sensitive [120]. Therefore evidence for IMTG being responsible for insulin resistance is shaky.
An interesting argument has been made to explain why individuals with abnormally low BMI are also frequently glucose intolerant [121]. Lipoatrophy, that is, absence of stored fat in the body is almost invariably associated with insulin resistance [122]. Diabetes associated with undernourishment is also reported [123]. The suggested explanation is related to the adipose tissue expandability hypothesis. The adipose tissue is said to sequester fatty acids and thus has a protective role against lipotoxicity [124]. In lipoatrophic condition, adipose tissue fails to develop, and as a consequence, FFAs keep on circulating in blood and cause insulin resistance. Another piece of evidence that adipose tissue by itself is not responsible for insulin resistance comes from transgenic mice overexpressing phosphoenolpyruvate carboxykinase (PEPCK) in adipose tissue. The enzyme PEPCK facilitates glycerol formation and esterification of FFAs into fat. Adipose tissue-specific overexpression of PEPCK increases fat mass and body weight without changes in circulating glucose, insulin, leptin, or FFAs. This model of obesity is not accompanied by insulin resistance demonstrating that the association of obesity with insulin resistance is not obligate. Rather than adipose tissue itself, something in the process of weight gain and fat accumulation could be causative to insulin resistance. The researchers who reported this finding think it is FFAs that induce insulin resistance, not adipose tissue [125]. Since FFA levels are unaltered in these mice, they remain insulin sensitive in spite of being obese. But also unaltered are leptin and insulin levels, raising the possibility that these might have a central role instead of FFAs.
The student looks too puzzled now. The arguments appear to be maneuvered according to short-term convenience of explanation. It looks as if the conclusion that obesity induces insulin resistance is predecided and details are maneuvered as and when needed in order to keep the conclusion intact. Whenever any evidence is found to contradict the original argument, the argument is maneuvered to explain the inconvenient truth. The new argument also faces some inconvenient truths later and therefore gets further maneuvered. While doing this, the earlier contradiction is often conveniently forgotten. When the mechanism by which FFAs induce insulin resistance is not found, adipose tissue and adipokines are blamed. When the paradox of adipokines is exposed, IMTGs are blamed which face the athletes’ paradox anyway. Since people with absence of adipose tissue are found to be insulin resistant, and there can be increased adipose tissue without insulin resistance, the ball is thrown back in the court of FFAs. Such surreptitious skipping between sub-hypotheses makes the main hypothesis unfalsifiable, and the student is certainly reluctant to believe in a theory that is unfalsifiable.
Summarily the relationship between obesity and insulin resistance is not as clean as presumed by the mainstream thinking. Obesity does not appear to meet the “necessary and sufficient” criteria for causation since we do find classes of individuals that are obese but insulin sensitive and also classes that are non-obese but insulin resistant. Also the mechanisms of obesity-mediated insulin resistance are not clearly elucidated so far. There are serious problems with every mechanism suggested so far. Therefore the causal role of obesity in insulin resistance is questionable although a statistical association cannot be denied. In brief, there are a number of gaps, contradictions, and possible flaws in the diet and obesity paradigm. This need not mean that diet and obesity are not important, but it certainly means that this line of thinking is missing something crucial and that “something else” could be more important than obesity itself.
3.Compensatory hyperinsulinemia: We will now examine the next postulate that β cells produce more insulin in order to compensate for the insulin resistance. As seen in the last chapter, in the HIIR state, there is a high level of insulin resistance as well as high levels of insulin in blood. However this is not sufficient to believe that hyperglycemia develops in order to compensate insulin resistance. Association is not a proof of causation. Following a good undergraduate classroom practice, alternative possibilities also need to be examined. The alternative possibilities are that (1) insulin overproduction is primary and since high levels of insulin can lead to hypoglycemia, insulin resistance develops as a compensating mechanism or (2) both are secondary to an unknown primary change that stimulates both the processes simultaneously and in a balanced manner.
If we assume that the primary process begins first and that there is a detectable lag in the response, the first to appear should be the primary cause, and the one to appear later should be the compensatory response. In the normal course of developing insulin resistance, both insulin resistance and hyperinsulinemia are observed simultaneously, and therefore, it is difficult to know what precedes and what follows. Fewer studies have followed the time course of development in sufficient details. In all these experiments, the first detectable change was a slight hypoglycemia [75, 126–129] indicating that hyperinsulinemia was stronger than insulin resistance in the beginning. Neonatal hypoglycemia is extremely common in low birth weight babies [130] that have a high propensity to become insulin resistant later. The early transient hypoglycemic phase in the natural history of T2D is known for a long time. The early hypoglycemic phase was one of the strong supporting pieces of evidence that Neel based his thrifty gene argument on (discussed in details in the following chapter). Neel’s arguments are actually based on the assumption that hyperinsulinemia is the starting point of diabetes [75]. It was somewhat later that the followers of thrifty gene theory painted the picture upside down.
But early hypoglycemia by itself may not be considered a conclusive evidence of hyperinsulinemia first. Alternative explanations are possible. For example, it can be argued that the compensatory reaction from pancreas comes instantaneously without any detectable lag and initially the reaction is stronger than needed and finds the right level of response iteratively during which transient hypoglycemia may be observed. A better way to resolve between the alternatives would be to selectively induce one of the processes and see the response of the other. Such situations do exist naturally or experimenters have achieved such feats. In certain types of pancreatic tumors, insulin is overproduced. This overproduction is due to the tumor and therefore can be safely assumed to be primary. An insulin producing tumor is always accompanied by insulin resistance, and if this tumor is removed, insulin sensitivity increases [131–136]. Therefore insulin resistance appears to be the compensatory response to hyperinsulinemia in this case implying that mechanisms do exist by which insulin resistance can develop to compensate hyperinsulinemia.
A reverse experiment in which insulin resistance was artificially induced and therefore we certainly know it was primary is also available. The muscle tissue is responsible for majority of insulin-dependent glucose uptake in the body. If insulin receptors in the muscle are specifically knocked out (such mice, called MIRKO for muscle insulin receptor knockout, are useful models of muscle insulin resistance), making the muscle tissue completely insulin resistant, insulin levels remain surprisingly normal [137]. It appears therefore that at least muscle insulin resistance is not compensated by hyperinsulinemia. A whole body insulin receptor knockout is nevertheless hyperinsulinemic [78]. So there is something else other than muscle insulin resistance that increases the insulin response. One can think of a possible reason that could bring about this change. If insulin exerts a negative feedback on its own secretion and β cell insulin receptors are needed for this action, insulin resistance at the β cell receptor level could result in hyperinsulinemia. But contrary to our expectation, β cell-specific insulin receptor knockouts are deficient in insulin production [138]. It can be concluded therefore that β cell insulin resistance cannot result in hyperinsulinemia. On the other hand, liver-specific insulin receptor knockouts do result in hyperinsulinemia, but this is accompanied by hyperglycemia, and therefore, here insulin production could be simply triggered by hyperglycemia [77]. It appears from these studies that compensatory hyperinsulinemia does not appear to follow insulin resistance in muscle and other tissues unless there is hyperglycemia. Therefore a normoglycemic HIIR state cannot be explained by the compensatory hyperinsulinemia hypothesis.
In contrast with insulin receptor knockouts, knockouts for the insulin receptor substrate (IRS-1 and IRS-2) become hyperinsulinemic [77]. IRS-1 knockouts are hyperinsulinemic in the absence of hyperglycemia. This is an example where induced insulin resistance is accompanied by compensatory hyperinsulinemia. But there is an alternative interpretation. IRS-1 and IRS-2 are also involved in insulin-like growth factor-1(IGF-1) signaling, and IGF-1 deficiency has been shown to lead to hyperinsulinemia and insulin resistance [139]. So in this case, IGF-1 resistance rather than insulin resistance could be responsible for hyperinsulinemia. As yet, there is no demonstration of induced impairment of insulin signaling that increases insulin response without involving IGF-1. Therefore compensatory hyperglycemia in response to specific impairment of insulin signaling in the absence of hyperglycemia is yet to be clearly demonstrated.
Intrauterine growth retardation is known to lead to insulin resistance and hyperinsulinemia in early life. In some of the models of IUGR, the development of hyperinsulinemia and insulin resistance has been studied in great detail. The FASDEL is such a model in which there is heterozygous deletion of fatty acid synthase. The FASDEL females give rise to small-sized babies owing to retarded intrauterine growth. In the FASDEL model of IUGR mice, at 3 months of age, there is larger islet area and β cell mass accompanied by hyperinsulinemia but normal insulin sensitivity leading to hypoglycemia. However at 12 months of age the picture is reversed. Older mice are glucose intolerant, hyperglycemic, and relatively insulin deficient indicating that hyperinsulinemia appears first in the sequence [140]. The early hyperinsulinemia in this model is certainly not induced by insulin resistance because insulin resistance is not demonstrable till a much later stage. A number of other studies also show a primary occurrence of hyperinsulinemia independent of insulin resistance [128, 141–142].
Let us also look at the mechanisms available that bring about the respective compensatory responses in either case. If pancreas has to compensate insulin resistance, they need to know the level of insulin resistance of the body and accordingly increase the insulin production level. The predominant mechanism that according to the classical theory stimulates insulin production is raised blood glucose. But ironically, the blood sugar is not raised in the early insulin-resistant state. In fact, in the early hyperinsulinemic phase, there could be slight hypoglycemia. Therefore insulin production does not appear to be stimulated by glucose. No other mechanism has been suggested even speculatively that can measure the level of insulin resistance and instruct the β cells to produce appropriate amounts of insulin.
On the other hand, many pathways exist by which insulin resistance can be induced when β cells overproduce insulin. (1) Insulin itself downregulates insulin receptors and desensitizes postreceptor pathways [143]. (2) A negative feedback loop by which insulin action can be regulated appears to be built in the system in the form of the mTOR/S6K1 pathway [42]. Insulin signaling activates mTOR, and mTOR with its downstream S6K1 signaling reduces the insulin receptor substrate response. As a result, if insulin signaling increases unduly, its effect gets downregulated automatically. (3) Another known pathway operates through amylin. Amylin is coproduced by the β cells along with insulin. Whenever the β cells are hyperactive and secrete excessive insulin, they also secrete excessive amylin. Amylin in high concentration has been well demonstrated to induce insulin resistance [144–150]. So the mechanism to avoid the risk of acute hypoglycemia if and when insulin is overproduced is built in the pancreas itself. (4) Alternatively, insulin induces the release of soluble Klotho protein from the membrane-bound form [151], and Klotho induces insulin resistance [152, 153]. (5) Raised levels of insulin stimulate serotonin secretion [154] in the brain, and chronically increased serotonin signaling in the hypothalamus is also known to induce peripheral insulin resistance [155]. (6) Insulin is known to stimulate lipogenesis, and both plasma triglycerides and adipose fat are implicated causatively in insulin resistance. Suppressing insulin production by somatostatin and its analogues is known to reduce obesity [156] and thereby insulin resistance. Therefore if insulin levels are raised for sufficiently long duration, insulin resistance should automatically follow. Out of the above mechanisms, (1)–(4) are relatively quick-acting mechanisms, and (5) and (6) have chronic actions. At this stage, we do not clearly know the relative importance of the alternative mechanisms. The fact of relevance here is that many mechanisms for compensatory insulin resistance exist, whereas those for compensatory hyperinsulinemia have been elusive.
All the evidence together raises doubts about whether hyperinsulinemia is an attempt to compensate insulin resistance or vice versa. Evidence is clearly in favor of the latter, that is, hyperinsulinemia is a primary response and insulin resistance arises as a compensatory reaction to hyperinsulinemia. If hyperinsulinemia comes first, the logical links between postulates 1–2 and 2–3 are in trouble and postulate 4 just cannot be true. If hyperinsulinemia comes first, β cell degeneration cannot be said to be the cause of failure of compensation, and if that is so, what makes the blood sugar go up? This could be a fatal blow to the classical thinking. Not surprisingly, there is extreme reluctance to accepting or even considering the idea that hyperinsulinemia could be coming first. This is not being said for the first time. In recent years, a number of authors have suggested this possibility giving evidence [75, 157, 158]. But it does not seem to have made a major impact on the mainstream thinking. The teacher in me is more conservative. Although hyperinsulinemia first is sufficient to make the entire theory of diabetes collapse, he convinces the student that it is prudent to ignore this for the time being and continue examine the rest.
If we accept that hyperinsulinemia comes first, we need to worry about what causes hyperinsulinemia. The orthodox theory does not offer any suggestion on this since there has been a mind block on “insulin resistance first.” Recently there have been suggestions as to why primary hyperinsulinemia develops in diabetes-prone individuals, and we will discuss them in subsequent chapters. For postulates 1 and 2, we only raised doubts, but for postulate 3, it appears to be a clear case of the textbook being wrong. But if postulate 3 is wrong, there are implications for 1, 2, and 4 as well.
There are a series of horse and cart paradoxes in the current understanding of obesity and diabetes in which the cause–effect relationships are terribly messed up. This is the first of the series. There are many more to come below. We will have to resolve all these paradoxes ultimately. I will call the paradox of whether insulin resistance first or hyperinsulinemia first as the horse and cart paradox I (one) following Shanik et al. [157].
4.Beta cell exhaustion/defect/failure resulting in relative insulin insufficiency leading to hyperglycemia: The next postulate is that of a relative deficiency of insulin causing hyperglycemia. In an early insulin-resistant individual, there is high level of insulin resistance, but glucose level remains normal since the higher levels of insulin are supposed to compensate it. Now since we are examining the current paradigm, let us forget for the time being that hyperinsulinemia comes first. We will continue to follow and examine the classical line of thought. It is believed that by having to produce large amounts of insulin over a long time, the β cells get exhausted and some kind of defects in β cells start accumulating. Histologically progressive islet degeneration can be evidently seen in T2D and is remarkable in the later stages but could be beginning very early in the history of T2D. Therefore there is no doubt that β cell degeneration does happen. The debate is whether exhaustion is the cause of this degeneration and whether the degeneration is a necessary and sufficient cause of hyperglycemia. Doubts about the β cell exhaustion theory have been raised from the beginning. Neel himself was quite unhappy about the concept of organ exhaustion and has refuted the claim [75].
The idea of chronic fatigue of any organ in the body is quite strange. As a teacher, I like to give analogies to illustrate a point. The problem in this case is that I can find no other example of chronic fatigue. Acute fatigue is quite well known but there is no good example of chronic fatigue. For example, if you run for 5 km at a stretch, your muscles will certainly experience fatigue. This is acute fatigue. But if you start running 5 km every day, then the muscle actually becomes stronger and stops getting exhausted. Except where physical wear and tear is involved such as teeth, this is the typical response of any organs to chronic overuse. Chronic overuse strengthens rather than weakens an organ. Even in the pancreas, there is an increase in the β cell mass in the early HIIR state. Apart from the belief associated with pancreatic islets, there is no other example of any endocrine organ failing or degenerating due to overuse. The adrenal cortex is hyperactive in chronic stress, but there is no demonstration of exhaustion. Thyroid hypertrophy during pregnancy or goiter is well known, but this is not known to be followed by degeneration of the endocrine gland. Still islets are believed to degenerate by chronic overwork.
A knockout experiment helps us to resolve this question once again. IRS-1 deficiency is known to induce insulin resistance and hyperinsulinemia simultaneously but does not directly affect β cell function. In IRS-1 knockouts, diabetes never results in spite of long-standing hypersecretion of insulin. Under this condition, β cells can keep on hypersecreting without getting exhausted. On the other hand, IRS-2 deficiency, in addition to inducing insulin resistance, directly affects β cell development and survival by interfering in the IGF-1 action on β cells [77]. Needless to say, these mice become diabetic. This is a demonstration that hyperinsulinemia by itself does not lead to β cell degeneration and cause diabetes. There needs to be another independent cause of β cell degeneration to develop hyperglycemia. Again there is this mystic “something else.”
The organ exhaustion theory is rather old, and now we know many other possible reasons for islet degeneration [159]. They include oxidative stress, glucotoxicity, lipotoxicity, and amyloid deposition, some of which are discussed below. Whether due to exhaustion or any other reason, there is no doubt that both insulin resistance and β cell dysfunction characterize type 2 diabetes. But the most relevant question that we need to pose is whether a combination of these two is both necessary and sufficient to explain fasting and postprandial hyperglycemia and further other complications of diabetes. This question is more important than what causes β cell degeneration. Therefore let us try to handle it first and then return to the causes of β cell degeneration. Overt diabetes is believed to appear when islet degeneration begins. Some kind of defect develops in β cells which are unable to give a compensatory response to insulin resistance.
How can we test this postulate? A simple way to test would be to mimic the hypothetical process and see whether the result is the same. For example, let us take a normoglycemic HIIR individual. Now, if insulin production in this individual is artificially and specifically suppressed, sugar levels should go up. If we can demonstrate this, we will be able to demonstrate that insulin resistance and insulin insufficiency are “sufficient” to explain hyperglycemia.
Such experiments have actually been done repeatedly. The big surprise of these experiments is that when insulin production is suppressed, the insulin sensitivity increases rapidly such that the sugar level remains more or less unaffected. Diazoxide is known to suppress insulin production. In genetically obese Zucker rats, the suppression of insulin production by diazoxide increased insulin sensitivity so much that the fasting sugar levels reduced instead of increasing. Further there was weight loss, increased fat oxidation, and improved lipid profile as well [160–162]. The effect of insulin suppression in humans was of a similar nature. Diazoxide treatment resulted in insulin suppression and simultaneously improved insulin sensitivity [158]. In another study, insulin suppression by diazoxide increased blood glucose levels in non-obese subjects but increased insulin sensitivity without altering glucose in obese subjects [163]. Alternatively insulin can be suppressed by protein deprivation. Using this dietary means to suppress insulin, not only insulin levels decreased but even glucose levels decreased [46], indicating that insulin suppression immediately increased insulin sensitivity. Another insulin suppressing agent octreotide had similar effects in human trials [164–166]. A combination of insulin siRNA and insulin degrading enzyme was able to produce diabetes-like condition in transgenic mice at high levels of suppression of insulin, but my analysis of their data showed that insulin sensitivity actually increased in the insulin-suppressed mice. About 75% of suppression of insulin production was needed to produce a detectable impairment of glucose tolerance [167]. Consistent demonstration of insulin sensitizing effects of insulin suppression by three different pharmacologically active agents as well as by dietary modification makes it highly unlikely that insulin sensitivity was an inadvertent effect of the agent itself. Suppression of insulin in an insulin-resistant state not being able to produce hyperglycemia is what I would like to call horse and cart paradox II. We made the horse move in the direction of diabetes by suppressing insulin but the cart did not. Instead of raising blood sugar, suppression of insulin appears to increase insulin sensitivity for an individual in an insulin-resistant state. The most logical interpretation of these “inconvenient” results is that when insulin production is suppressed, insulin resistance actually seems to adjust itself to keep the sugar levels normal. This compensatory adjustment in insulin sensitivity can fail only when the insulin levels become too low. If we have accepted that hyperinsulinemia is primary and insulin resistance is a compensatory response, then this result is not a surprise at all. If insulin levels are high, insulin resistance is required by the body; otherwise, the sugar levels will drop down to dangerously low levels. But if insulin levels are brought down artificially, then insulin resistance is no more needed, and it comes down to such a level that sugar remains more or less normal.
In these experiments, insulin-resistant state already existed and we suppressed insulin production, so following the classical paradigm, we would have expected the blood sugar level to rise. But the blood sugar level did not go up, indicating that insulin insufficiency on the background of insulin resistance is not “sufficient” to cause hyperglycemia.
Another experiment that raises doubt about IR–RII being sufficient cause of hyperglycemia is again that of MIRKO mice. In these mice, insulin signaling in muscle is reduced by over 95%. This is an extreme possible case of muscle insulin resistance. By classical thinking, we would expect insulin levels to increase in compensation, but they do not as seen earlier. Now if there is extreme muscle insulin resistance and no compensatory insulin production, it must be an ideal situation for blood glucose to go up. But most surprisingly, that does not happen. Sugar levels remain normal in spite of extreme insulin resistance and lack of compensatory rise in insulin [77]. This means that extreme insulin resistance and lack of compensatory insulin response were not “sufficient” to increase blood sugar in MIRKO mice. How did it happen? In the previous experiment, there was an increase in insulin sensitivity when insulin production was suppressed. Here since the insulin receptor itself is knocked out, this possibility is ruled out. There are two other compensatory mechanisms which evidently work to compensate effectively for both insulin resistance and lack of compensatory increase. One is a compensatory glucose uptake by tissues that have intact insulin receptors. Evidence for this comes from the observation that fat cells of MIRKO mice take up excess glucose and increase their fat stores. The other mechanism is that muscles start making use of insulin-independent pathways for picking up glucose. Muscle glucose uptake during exercise is independent of insulin, so insulin-independent pathways do exist. This is evident by the observation that there is no wasting of muscle in MIRKO mice which is expected if insulin action in muscle is completely stopped and not compensated by any other mechanism. The other evidence is that muscle-specific glut-4 knockouts do become hyperglycemic in contrast with MIRKO. This is because ablation of glut-4 blocks the insulin-independent pathways as well [168]. The important insight obtained from these knockout experiments is that mechanisms exist in muscle tissue that can compensate for insulin resistance, but for some strange reason, they do not seem to work in diabetes. Which again points to “something else” being responsible for diabetic hyperglycemia in addition to IR–RII.
Another example of insulin-independent mechanisms compensating for insulin resistance comes from liver-specific insulin receptor knockout (LIRKO) mice. LIRKO mice were observed to be severely insulin resistant as well as hyperglycemic and hyperinsulinemic. We have seen in the last chapter that fasting hyperglycemia mainly reflects liver insulin resistance. As expected, young LIRKO mice had fasting hyperglycemia. However, by 6 months of age, their fasting sugar levels returned to normal without gaining liver insulin receptors back or increasing insulin secretion further. At 2 months, the mean fasting glucose of LIRKO was 132 mg/dl which reduced to 70 mg/dl by 6 months, but the fasting insulin levels remained the same [169]. Since there was no possibility of increasing insulin sensitivity of the liver, we need to infer that some other compensatory mechanism restored the control on liver glucose metabolism resulting in normalization of fasting blood glucose. This example is very dramatic. Completely knocking out the insulin receptor means not only that there is insulin resistance but also that increasing insulin levels cannot compensate for the loss. But in spite of this, fasting sugar levels returned to normal. This means that some compensatory mechanisms do exist which are independent of insulin. Liver glucose production is known to be directly under the control of hypothalamic regulation through the vagus nerve. When insulin mechanism fails to work, this mechanism appears to take over the control of the situation and normalize metabolism. So if insulin signaling in the liver fails, there are compensatory mechanisms to take care. But surprisingly, these compensatory mechanisms appear to keep mum during diabetes. This is one more demonstration that something other than IR–RII is happening in diabetes.
The unanswered question now is what exactly happens in diabetes. By the current belief, in an insulin-resistant state, whenever compensatory insulin production is not adequate, the blood sugar goes up. This is not different from the above experiments in which there were both insulin resistance and lack of compensation by insulin. But what happened in the experiments does not match with what happens in diabetes. The mechanism of adjusting insulin sensitivity to the current insulin production or that of compensating insulin resistance by insulin-independent pathway fails when diabetes sets in. Since insulin resistance and insulin suppression were unable to raise blood sugar, we suspect that some factor other than these two must be responsible for the diabetic hyperglycemia. This something else must be more important and central to T2D than the combination of insulin resistance and insulin decompensation. Possible answers to this question will come later in this book. Here it is sufficient to make a statement that IR–RII is not “sufficient” to cause a rise in blood sugar.
We can start experiments from the other end now to ask whether IR–RII is “necessary” for hyperglycemia. One approach would be to look for natural or experimentally induced cases where hyperglycemia or impaired glucose tolerance is seen without insulin resistance or without β cell degeneration. Two such natural situations exist. One is a condition called anorexia nervosa, generally affecting women and characterized by obsessive fear of becoming fat which adversely affects food intake. As a result, patients with this condition are underweight and scarcely have fat. Although they are highly insulin sensitive, they show impaired glucose tolerance very frequently. Since there is no evidence of β cell degeneration in anorexia nervosa [170], it appears that there can be impaired glucose tolerance in the absence of both insulin resistance and β cell degeneration [171]. Interestingly, anorexia nervosa patients also have high cholesterol despite low calorie intake, and their cholesterol can be normalized by increasing calorie intake and body weight [172–174]. The other example comes from a small study on the Kalahari !Kung which showed that the tribesmen were insulin sensitive but still had impaired glucose tolerance [73] by the Western standards.
I can perceive another experiment where we should take individuals with mild fasting hyperglycemia which is presumably caused by IR–RII. If the popular picture that β cells are trying to keep the glucose levels normal by producing as much insulin as they can but they have reached their limit and are unable to compensate sufficiently for the level of insulin resistance is correct, then we would expect that after bringing down insulin resistance by exercise, the compensation should work and glucose levels should come down towards normal. Insulin levels may reduce following normalization of blood glucose. It is well known that exercise reduces insulin resistance. This has been shown reproducibly by a large number of studies using different measures of insulin resistance. All of them invariably show that exercises increase insulin sensitivity [175–181]. We also have effective insulin sensitizing drugs available. The question of my interest is whether insulin levels come down first or glucose levels come down first. If sugar levels reduce first and in response insulin levels drop, this is compatible with the current understanding. But if the reverse happens, it indicates that something different is happening. Published literature does not give a clear idea as to what comes down first. This is because the time course of events is either not observed or not reported with sufficient resolution. I happened to see some unpublished data in which in a substantial proportion of individuals participating in a trial, exercises reduced insulin resistance and insulin levels dramatically in a few weeks, but fasting sugar levels and HbA1c either did not reduce significantly or did so only after 10–12 weeks.
The question whether insulin comes down first or glucose does not seem to be addressed directly anytime. Nevertheless I will cite a few published reports which do not address this question but we can draw some inferences indirectly from their data. In a study, diabetic mice having fasting plasma glucose close to 9 mmol/l were treated with insulin sensitizing drugs, namely, metformin and rosiglitazone. After a year-long treatment, their glucose-stimulated insulin secretion decreased substantially, but glucose levels did not [182]. Another study compares the glucose tolerance curves of young versus sedentary old and exercising old individuals. As compared to the young age group, the sedentary old individuals have higher glucose levels as well as higher insulin levels in OGTT indicating insulin resistance. On exercise training, the insulin curve of older individuals reduced and became indistinguishable from those of the young. But glucose curves did not change significantly indicating that when insulin sensitivity increased, it was insulin that came down, not glucose [176]. In another study on individuals with obese postmenopausal diabetic women with basal average glycated hemoglobin of 6.9 (the normal range being 5.7–6.4), exercises increased the glucose disposal rates indicating increased insulin sensitivity, but HbA1c did not show any change [183]. Readers may wonder why I suspect that insulin may come down first and why I am specifically interested in this question. This will be answered in a later chapter. The point of relevance here is that if sugar levels remain unchanged even after reducing insulin sensitivity, it might indicate that there is something else that is determining sugar levels apart from IR–RII. Therefore this issue needs to be addressed with experiments specifically designed to answer this question. If we find that insulin levels respond more rapidly to insulin sensitizing treatment than sugar levels, that will make our horse and cart paradox III where we stopped the horse but the cart kept on moving. We reduced insulin resistance but hyperglycemia continued.
An inevitable conclusion of the multiple experiments above is that although insulin resistance and insulin insufficiency due to β cell degeneration are both undoubtedly involved in glucose homeostasis, the two are neither necessary not sufficient to cause hyperglycemia. Since the “necessary and sufficient” criteria are not met, it would be more appropriate to call insulin resistance and β cell degeneration as risk factors for hyperglycemia rather than causes of hyperglycemia. There is something missing, something perhaps more complex or more elusive that appears to determine the blood sugar levels, and both insulin resistance and insulin production appear to adjust themselves to maintain that mystic level. Two more sets of interesting experiments demonstrate that apart from insulin resistance and insulin insufficiency, there is something more involved in deciding blood sugar levels.
Do you think high plasma glucose levels can be reduced by infusing more glucose in the body? This sounds weird but it does happen provided glucose is infused in specific sites in the body. One such site is the portal vein. If glucose is infused directly in the portal vein which carries blood from the gut to the liver, plasma glucose levels drop rapidly [184]. Researchers performing these experiments claim that there is a portal glucose sensor that measures the glucose levels in the portal blood and the rate of change in these levels. Signals from these sensors go to the brain through sensory neurons. Rising levels of portal glucose quickly suppress gluconeogenesis by the liver and stimulate glucose utilization by muscle, both the processes happening apparently independent of insulin. This may result into hypoglycemia. Moreover, if the amount of glucose infused in the portal vein is calculated to match the amount of glucose that liver would have produced otherwise, liver glucose production completely stops [184]. This is as if the liver was assigned a quota for glucose production by someone, and since the liver was then fooled to think that the required amount of sugar was already coming from the gut, it completely stopped producing glucose.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


