(1)
Indian Institute of Science Education and Research Pune (IISER-P), Pune, India
Abstract
Before we start thinking about the evolutionary origins of diabetes and related disorders, I need to briefly sketch what is currently known and well accepted about diabetes. This chapter tries to compile a textbook picture of diabetes [1–3] only to serve as a background. Readers who have studied physiology or medicine may skip this chapter straightaway since it does not contain any new argument. It may be necessary and useful for readers who need a fair amount of background information about diabetes before appreciating the paradoxes and puzzles associated with it.
Before we start thinking about the evolutionary origins of diabetes and related disorders, I need to briefly sketch what is currently known and well accepted about diabetes. This chapter tries to compile a textbook picture of diabetes [1–3] only to serve as a background. Readers who have studied physiology or medicine may skip this chapter straightaway since it does not contain any new argument. It may be necessary and useful for readers who need a fair amount of background information about diabetes before appreciating the paradoxes and puzzles associated with it.
As a student and then as a teacher, I always found textbooks very boring. Since this chapter is an extract of textbooks, it would sound boring too. There is a class of readers that can cope with it. But if I were to be the reader, I would have given up reading the book midway in Chap. 2. To avoid this disaster I would suggest an alternative reading plan. The background textbook information in this chapter is needed in order to understand the questions raised and the stepwise arguments made in further chapters. But it is not necessary to read it right away. So for impatient readers like me, I would suggest skipping this chapter at whichever point it gets boring but returning to it as and when a reference to the basic textbook information is needed. Science becomes lively when one goes beyond textbooks. This can be achieved during lively discussions in a classroom, and that is the approach from the next chapter which I can assure is very different from this chapter.
So here begins the textbook stuff.
Diabetes is among the most widely known and also among the least understood of all disorders. It should also be listed among the earliest known disorders in the history of medicine. We find a description of some of the signs and symptoms of diabetes in ancient Egyptian and Indian literature. Polyuria or frequent and copious urination was among the first symptoms that we find recorded in Egyptian literature over 1500 BC. Indian medicine had noted that the urine of diabetics tasted sweet and would attract ants. By the middle of the eighteenth century, it was shown that the sugar levels in the blood of diabetics were raised substantially. A major contribution in our understanding of diabetes came from Claude Bernard who discovered a number of fundamental principles in physiology such as storage of energy by the liver in the form of glycogen. He demonstrated that damage to medulla oblongata caused severe hyperglycemia and therefore thought that brain was the main center for regulation of blood glucose [2, 4]. However, by the turn of the nineteenth century, it was shown that the removal of pancreas leads to diabetes. Soon the islets of Langerhans were described, and some soluble substance produced from there was suspected to regulate blood sugar. The name “insulin” was first used for this magic molecule in 1909, and by 1921, insulin was purified by Banting, Best, Collip, and Macleod. Almost simultaneously glucagon, the counter signal in glucose regulation, was being discovered. Glucagon was named in 1923. By then Claude Bernard’s demonstration of brain damage affecting glucose regulation was almost completely forgotten, and glucose levels were thought to be maintained by the balancing actions of insulin and glucagon.
Insulin, which is believed to be central to diabetes, is a peptide hormone secreted by β cells of pancreas in response to blood glucose levels, amino acids, and nervous stimulation. It is central to regulating carbohydrate and fat metabolism in the body. Normal insulin secretion responses are essential for maintaining normal body functions. Insulin has a wide variety of effects on different tissues of the body. In the adipose tissue it increases glucose uptake, fatty acid synthesis, glycerol phosphate synthesis, triglyceride deposition, activation of lipoprotein lipase, and inhibition of hormone-sensitive lipase. In muscles it increases glucose uptake, glycogen synthesis, K+ uptake, amino acid uptake, and protein synthesis. Therefore insulin action is important in muscle building and muscle strength. Insulin has an osteoblastic, i.e., bone-forming, function and thereby contributes to bone strength too. In the liver it decreases gluconeogenesis and ketogenesis and facilitates glycogen synthesis, glycolysis, protein synthesis, and lipid synthesis. More generally in the body, insulin is primarily a growth hormone promoting cell growth and protein synthesis. The growth-promoting function of insulin is synergistic with another hormone, the pituitary growth hormone (GH). Insulin has also been shown to be important in the cognitive functions of the brain. Insulin acts as the signaling molecule simultaneously regulating different functions of the body in different ways [3]. Therefore it is logical to expect that when there is some or the other type of problem in insulin function, not only blood sugar is affected but a large number of functions of the body are.
Before understanding the altered patterns of insulin secretion, normal patterns need to be understood. Glucose is a major stimulus for insulin secretion by pancreatic β cells. Insulin production is also stimulated by some other nutrients including amino acids such as arginine. Several other hormones of the body affect basal or glucose-stimulated insulin secretion (Table 2.1). In addition parasympathetic nervous system has a direct control over insulin secretion, but the significance of it in normal or pathological conditions is poorly understood. Under normal conditions out of total insulin secretion in 24 h, ∼50% is secreted under basal conditions while the rest is secreted in response to meals. The amount of insulin secreted for different types and timings of food intake in a day such as breakfast, lunch, and dinner does not differ significantly. Insulin secretion is not continuous but is oscillatory in nature. Three types of oscillations have been observed, namely, rapid small amplitude oscillations, ultradian oscillations consisting of large amplitude, slow oscillations, and circadian oscillations in the form of different insulin responses at different times of the day. The exact physiological significance of the oscillations is not clear, but exogenous insulin is more effective in reducing plasma glucose levels when administered in oscillatory doses than at a constant rate.
Table 2.1
Factors affecting insulin secretion
Stimulators | Inhibitors |
|---|---|
Glucose Mannose Amino acids Intestinal hormones [gastric inhibiting peptide, glucagon-like peptide, gastrin, secretin, cholecystokinin (CCK)] β-Keto-acids Acetylcholine Glucagon Cyclic adenosine monophosphate (cAMP) Β-Adrenergic stimulators Theophylline Sulfonylureas | Somatostatin 2-Deoxyglucose Mannoheptulose α-Adrenergic stimulators (epinephrine) Galanin Diazoxide K+ depletion Alloxan Insulin |
The insulin response to glucose or a meal has two distinct phases. The first is an acute phase where a brief peak of insulin secretion arises immediately after feeding, even before absorption of food begins. The acute phase insulin response (AIR) is driven by neuronal mechanisms and has sometimes been called cephalic phase insulin response. The second phase appears to be predominated by peripheral mechanisms involving islet stimulation by glucose. The acute phase response is important for the normal glucose tolerance curve. Absence of acute phase response increases the area under the curve for both glucose and insulin. AIR is impaired in diabetes from an early stage.
Diabetes and Its Types
Although insulin has a large number of functions, the definition and diagnosis of diabetes are based on only one of its functions which is regulation of peripheral blood glucose levels. Insulin is a signal molecule, and a signal can function well when it is given out as well as received normally. Endocrine signals are like radio broadcasting where the signal is given globally, but it can be received only where the specific receiving mechanisms exist. Such a signal will fail if either the mechanisms of relaying the signal or the mechanisms of receiving it are impaired. In type 1 diabetes the former is impaired and in type 2 the latter. Defective secretion of insulin causes type 1 diabetes, whereas defective mechanisms of insulin signal reception and downstream action appear to be the root cause of type 2 diabetes. Type 1 diabetes (T1D) most commonly results from autoimmune destruction of insulin-producing β cells of the pancreas. The subsequent lack of insulin leads to increased blood glucose and urine glucose. Although blood and urine glucose are the presenting signs of T1D, most of the pathology of untreated T1D results from deficiency of insulin rather than raised blood sugar.
Type 2 diabetes mellitus (T2D) is classically characterized by high blood glucose which is believed to be a result of a combination of insulin resistance and relative insulin insufficiency. We are going to refer to this condition frequently in this book and will call it as IR-RII throughout. The reduced response of tissues to insulin signaling is called insulin resistance. When insulin resistance sets in, higher amounts of insulin are needed to bring about the same function, just as shouting in louder voice is needed while talking to a hearing impaired. This is what is believed to happen in the body. Insulin resistance is a measurable character and is measured by different techniques tabulated in Table 2.2. The different indices capture somewhat different elements of insulin resistance but generally are intercorrelated sufficiently well. Clinically the most frequently used index of insulin resistance is called HOMA-IR which is proportional to the product of fasting insulin and fasting glucose levels. HOMA-IR reflects muscle as well liver insulin resistance. The gold standard of measuring insulin sensitivity is achieved by using a technique called euglycemic hyperinsulinemic clamp. In this technique insulin is infused continuously to achieve a high and stable plasma level. Because of high insulin level, plasma glucose drops down. The normal levels of glucose can be brought back by infusing glucose. At the stable high insulin level, the amount of glucose infusion needed to retain normal glucose levels is a measure of insulin sensitivity. This measure mainly reflects glucose uptake by muscle by the insulin-dependent pathways. The clamp technique is too complex to be used in clinical practice and is restricted to physiological research.
Clinically useful methods | Formula | Comments | |
|---|---|---|---|
Methods based on fasting measurements | Fasting insulin | I 0 | Simple and feasible methods in a clinical setup. More variability. Do not differentiate between liver and peripheral insulin resistance. Not useful in advanced diabetics |
HOMA | I 0 × G 0/constant | ||
QUICKI | 1/[log(I 0) + log(G 0)] | ||
G/I | G 0/I 0 | ||
Methods based on OGTT | Cederholm and Wibell | M/G × log I | More elaborate and reliable but more difficult to perform. Correlated well with clamp methods in normal prediabetics and early diabetics but weakly in advanced diabetics |
Matsuda et al. index | 10,000/√(G 0 × I 0) × (G × I) | ||
Insulinogenic index | (I 30 − I 0)/(G 30 − G 0) | ||
Many others | |||
Methods for research | |||
Clamp methods | Hyperinsulinemic euglycemic clamp | The quantity of exogenous glucose needed to maintain normal glucose under an induced stable hyperinsulinemic condition | The gold standard of insulin resistance measurement. Highly elaborate and tricky to perform but more reliable and reproducible. Differentially and specifically represents insulin-induced glucose disposal. Physiologically unrealistic conditions |
FSIVGTT | Frequently sampled intravenous GTT | Glucose clearance rate per unit change in plasma insulin concentration | Needs over 30 samples in a GTT. Estimates based on a computational model. Physiologically more realistic than clamp |
Insulin resistance is almost always accompanied by higher levels of insulin production such that the resultant function remains normal. This is believed to be because of a compensatory rise in insulin production in response to insulin resistance. High insulin resistance accompanied by raised level of insulin such that blood sugar remains normal is a state that is called insulin-resistant state which generally precedes T2D by several years. We will call the hyperinsulinemic insulin-resistant state as HIIR in which glucose levels are normal. Not all HIIR individuals develop T2D, but many do. This is believed to happen when the β cells are unable to compensate insulin resistance resulting into a relative insulin deficiency. This is said to be because of fatigue or exhaustion of the insulin-producing β cells of the pancreatic islets. At this stage the total insulin production of a diabetic may be higher than that of a nondiabetic and non-insulin-resistant individual, but blood sugars are raised owing to even higher levels of insulin resistance. Thus it is a combined effect of insulin resistance and relative insulin insufficiency (IR-RII) that is believed to lead to diabetes. T2D is the predominant form of diabetes worldwide, constituting about 90% of cases globally. The diabetes epidemic is increasing in both developed as well as developing countries. Globally the number of people with diabetes is expected to rise to above 300 million in 2015 [6], but this is an underestimate because, for each diagnosed case, there is thought to be one undiagnosed case in First World countries and eight in the Third World. Type 2 diabetes has become the world’s most important public health problem [6]. The focus of this book is on T2D, although while talking about pathophysiology of diabetic complications, where there is much overlap between T1D and T2D, we will include T1D in the discussions. In some of the older literature, T2D has been often referred to as “mild” diabetes. In the past 40 years, the status of T2D has changed from being considered as a mild disorder of the elderly to one of the major causes of morbidity and mortality increasingly affecting the middle aged and even youth. It is important to note that the rise in prevalence is seen in all six inhabited continents of the globe [7, 8].
The diagnosis of diabetes rests on the measurement of plasma glucose levels. Because plasma glucose concentrations range as a continuum, the criteria are based on estimates of the threshold for the complications of diabetes. Reproducibility of the plasma glucose concentration is an important issue for interpreting the results of diagnostic tests for diabetes. There is significant variation in the results of repeated tests in the adults after 2–6 weeks interval. Thus it is essential that any test showing above normal levels be confirmed by repeated tests. Before overt diabetes appears, alterations in glucose homeostasis are detectable if a curve of changing glucose levels after meals or glucose intake is monitored (Fig. 2.1). This can be done using what is called an oral glucose tolerance test. Although oral glucose tolerance test (OGTT) is an invaluable tool in research, it is not recommended for routine use in diagnosing diabetes. Levels of glycated hemoglobin A1c (HbA1c) are generally not used for diagnosing diabetes but can be valuable for follow-up. Hemoglobin along with many other proteins get glycated very slowly by spontaneously combining with glucose, and this glycation is directly proportional to the levels of glucose in blood. That makes HbA1c a cumulative estimate of glucose, averaging out the daily fluctuations in glucose levels. Individuals who deviate from the normal glucose tolerance curve but are not overtly diabetic are placed in an intermediate category called impaired glucose tolerance (IGT). The thresholds for the definitions are in Table 2.3.
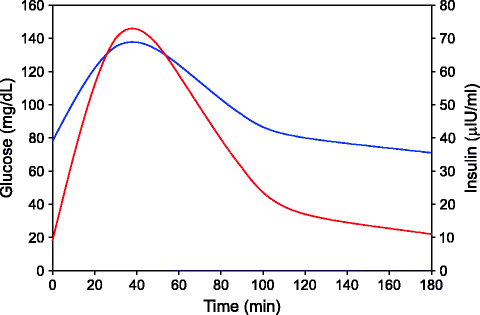
Fig. 2.1
A typical time course of plasma glucose and insulin levels after oral administration of 75 g glucose (oral glucose tolerance test OGTT). The nature of the curve reflects in multiple ways the state of glucose homeostasis in the body (Glucose is shown by blue line on primary Y axis, insulin in red on secondary Y axis)
Table 2.3
Diagnostic criteria for stages of type 2 diabetes
Test | Normoglycemia (mg/dL) | IFG (mg/dL) | IGT (mg/dL) | Diabetes |
|---|---|---|---|---|
FPG | <100 | 100–125 | ≥126 mg/dL | |
2-h PG | <140 | 140–199 | ≥200 mg/dL | |
Random plasma glucose concentration | ≥200 mg/dL plus symptoms of diabetes |
Type 2 diabetes is traditionally thought to have a strong genetic component. However the increasing incidence of the disorder cannot be of genetic origin since gene(s) that give rise to a disorder will not increase in frequency in a species globally. Even if it does, no allele can increase in frequency dramatically within only a couple of generations. So genes alone cannot be blamed for diabetes. The current thinking is that it is an interaction between genes and environment that is responsible for the current epidemic. Genetically predisposed persons who are exposed to a series of diabetogenic environmental influences develop clinical disease [9–11]. Although T2D is believed to have a strong genetic component, in exceptionally few cases, some specific genes are demonstrably involved. The common forms of diabetes appear to be polygenic in nature and are believed to be due to combination of genes involved in predisposing to obesity, insulin resistance, and abnormal insulin secretion.
The environmental and lifestyle factors predisposing to diabetes are sedentary lifestyle, altered diet leading to a positive energy balance, stressful life, excessive tobacco, and alcohol. In addition a strange pattern was discovered in the early 1990s that individuals born small for gestational age are highly predisposed to diabetes. This trend is consistently observed across the globe. Animal experiments have confirmed that intrauterine growth is crucial in determining the predisposition to diabetes. Intrauterine growth retardation (IUGR) leads to altered metabolic programming which makes a person prone to obesity and insulin resistance in later life.
Obesity and Muscle Insulin Resistance
The primary effect of insulin on glucose is to stimulate translocation of glucose transporters to cell membrane through which glucose enters into the cells and is then utilized for energy production. This happens through an elaborate cell signaling and downstream pathway. It appears that the number of glucose transporters in skeletal muscles of an insulin-resistant person is not different, but the ability of insulin to facilitate this translocation is disrupted. The term insulin resistance indicates an impaired biological response to either endogenously secreted or exogenously administered insulin. There is reduced insulin-mediated glucose transport and metabolism in skeletal muscles and adipocytes and also impaired suppression of hepatic glucose output. Insulin resistance is present in persons predisposed to type 2 diabetes before the onset of hyperglycemia, and so it has been concluded that insulin resistance is the primary abnormality that is responsible for the development of type 2 diabetes. Insulin resistance is present years before the onset of the disease, and this is a consistent finding in type 2 diabetes [12, 13]. A substantial amount of data indicates that insulin resistance plays a major role in the development of glucose intolerance and diabetes. Insulin resistance is associated with the progression to IGT and eventually to type 2 diabetes [14]. Prospective studies show that insulin resistance predicts the onset of disease. Insulin resistance is present in first-degree relatives of type 2 diabetics even when they are not obese. This is in support of having a strong genetic component in the etiology of T2D [12, 14, 15]. There is also a strong influence of environmental factors on the genetic predisposition to insulin resistance [16, 17]. The generally agreed pathway to T2D is that obesity leads to insulin resistance in genetically predisposed individuals which in the long run leads to T2D.
A number of studies show that the risk of insulin resistance and development of T2D increases with increase in body weight and particularly body fat content. It is not clear whether it is the free fatty acids in circulation or the adipose stores of the body that are of primary importance in inducing insulin resistance, but the general role of fat seems to be agreed upon. A close association between obesity and insulin resistance is seen in all ethnic groups and is found across the full range of body weights, across all ages, and in both sexes [18–20]. Absolute amount of body fat has an effect on insulin sensitivity across a broad range [21–23]. However central or visceral (intra-abdominal) adiposity is more strongly linked to insulin resistance, and accumulation of abdominal fat has effects on glucose tolerance independent of total adiposity [24–28]. The reason why intra-abdominal fat is more strongly related to insulin resistance is not clearly known at the proximate and ultimate level. There are a number of possible explanations for this strong association. It is speculated that due to presence of more number of adrenergic receptors as compared to subcutaneous fat, intra-abdominal fat is lipolytically more active [27, 28] generating more free fatty acids (FFAs) which induce insulin resistance. Another reason stated is that the intra-abdominal fat is more resistant to the antilipolytic effects of insulin [29]. One more hypothesis relies on the presence of high concentrations of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) that enhances conversion of inactive cortisone to active cortisol, leading to increased local cortisol production, which might be responsible for increased lipolysis. All these hypotheses appear to count on increased FFA production as the proximate cause of insulin resistance. A possible internal contradiction here is that if abdominal fat undergoes rapid lipolysis, it is difficult to account for greater accumulation of fat in the abdomen. Also obese persons have actually been shown to have lower rates of fat oxidation. Increased FFA level is related to insulin resistance in skeletal muscle. The mechanism by which FFAs reduce glucose transport has remained elusive. But it is shown that elevated free fatty acids (FFAs) predict the progression from IGT to diabetes [30, 31] implying some role for FFAs.
In contrast very low levels of lipids in the body or absence of adipose tissue, a condition called lipoatrophy, has a strong association with insulin resistance. So although fat is said to be responsible for inducing insulin resistance, the absence of fat also induces insulin resistance.
Alternatively intramuscular triglycerides (IMTG) rather than FFAs are implicated in insulin resistance. A strong correlation between IMTG concentration and insulin resistance has been demonstrated by evaluating IMTG with biopsy [32]. IMTG may accumulate if the rate of FFA uptake into the muscles and rate of their oxidation mismatch. Again although insulin-stimulated glucose uptake and the amount of IMTG are shown to be inversely related in nonexercising individuals, the mechanism by which IMTGs induce insulin resistance is not clearly known. Furthermore increased IMTG content is not invariably linked to insulin resistance because long-term exercise training is linked with increased IMTG [33], and chronic exercise increases insulin sensitivity as well as IMTG and the capacity for fatty acid oxidation [34–37].
A yet another possibility is that of a central role of malonyl CoA levels. Glucose uptake even in insulin-resistant muscle is higher at the elevated levels of blood glucose as seen in T2D [38, 39]. The resulting high glycolytic activity can generate acetyl CoA which is converted to malonyl CoA. The accumulation of malonyl CoA inhibits the carnitine palmitoyltransferase (CPT I) which resides on outer mitochondrial membrane, inhibiting uptake of acyl CoA [40]. The resulting buildup of acyl CoA and diacylglycerols is proposed to activate one or more protein kinases, resulting in insulin resistance [40]. Insulin-sensitizing effect of exercise supports this hypothesis since exercise lowers intracellular long-chain acyl CoA levels [41]. However, this pathway has a built-in negative feedback since there will be a decreased glucose flux after developing insulin resistance which will reduce the pool of acetyl CoA, eventually reducing malonyl CoA. Therefore this pathway may not induce sustained insulin resistance.
Higher nutrient status is known to activate the mammalian target of rapamycin (mTOR) pathway which through the S6 kinase 1 pathway ultimately decreases the levels of available insulin receptor substrate IRS1 and thereby induces insulin resistance. However insulin signaling itself is needed for expression of mTOR, and therefore, mTOR and insulin signaling appear to be in a negative feedback loop. Negative feedback loops are generally self-regulated and therefore do not lead to escalation. Therefore mTOR activation alone may not be sufficient to explain sustained insulin resistance.
The upcoming alternative explanation for the connection between obesity and insulin resistance is that the adipocytes secrete a number of signal molecules called adipokines which induce insulin resistance. In this line of thinking, the focus is shifted from FFAs to the adipose tissue. A wide variety of compounds that constitute adipokines are known, and their association with insulin resistance has been demonstrated. TNF-α appears to be the most important adipokine in this connection, and the pathway leading to insulin resistance appears to involve inflammatory signals, and yet the precise pathway of induction of muscle insulin resistance remains to be completely elucidated. Therefore although there is a strong correlative evidence for the role of free fatty acids, triglycerides or adipose tissue and its secretions in insulin resistance, and a number of possible pathways are visualized, the exact molecular events which lead to development of sustained insulin resistance in obesity are still not well understood.
There are a number of other possible mechanisms leading to insulin resistance which are not directly dependent on lipid metabolism. The brain monoamines such as serotonin and dopamine influence insulin sensitivity in a complex way, but their actual role in clinical insulin resistance is not clearly known. Insulin resistance is associated with decreased mitochondrial activity. Decreased mitochondrial activity may be responsible for decreased oxidative metabolism and eventually insulin resistance. But a reverse causation is equally possible. Insulin itself is important in upregulating mitochondrial biogenesis, so in an insulin-resistant state mitochondrial activity might be reduced. High-fat diet, although known to induce insulin resistance, was actually shown to increase mitochondrial mass in rats. Therefore, fat-induced insulin resistance is unlikely to work through decrease in mitochondrial activity [42]. The association of decreased mitochondria and insulin resistance is either because of reverse causation or some other causative factor. The central nervous system also plays a role in regulating hyperinsulinemia and insulin resistance. There is evidence for complex interactions between central and peripheral signals modulating insulin sensitivity and secretion [43–46], but the nature of these interactions is not yet understood sufficiently well.
In brief, although the concept of insulin resistance is central to type 2 diabetes, the causes and pathways leading to insulin resistance are not yet clearly known.
Liver Insulin Resistance
Insulin’s predominant action on liver is to regulate the hepatic glucose output. Liver has the important job of synthesizing glucose and releasing it into blood in fasting conditions. This is achieved by mobilizing the glycogen stores in the liver (glycogenolysis) or by making glucose from fatty acids or amino acids (gluconeogenesis). Liver glucose production is important during fasting since the brain is crucially dependent on glucose. The fat stores of the body are mobilized under starvation to release fatty acids. Most other tissues can directly use fatty acids as fuel but the brain cannot. Glucose production is therefore most essential for brain function in fasting conditions. However, the glucose production activity needs to be under regulation, and insulin works as a negative regulator of this activity. Impaired insulin signaling in the liver leads to defective regulation of liver glucose production. Although studies have suggested that kidneys can contribute to some endogenous glucose production, the defective glucose production in T2D is primarily in the liver. Role of nervous system and glucose autoregulation of hepatic glucose production is also known but is currently considered to be less important. Insulin decreases endogenous glucose production by direct and indirect mechanisms. In its direct action, portal insulin suppresses glucose production by inhibiting glycogenolysis. Insulin decreases gluconeogenesis simultaneously by directly inhibiting the enzymes required for gluconeogenesis and also through indirect routes by reducing the availability of free fatty acids required for gluconeogenesis and by inhibiting the secretion of glucagon that stimulates glucose production [47–51]. In diabetes insulin is not able to exert an impact on liver, so there is uncontrolled hepatic glucose production [52–54] which leads to raised fasting blood sugar. This is called liver insulin resistance. When the fasting glucose level is marginally high, the contribution is mainly from decline in glucose clearance by muscle and other tissues. However, higher levels of fasting glucose are mainly contributed by hepatic glucose production.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


